
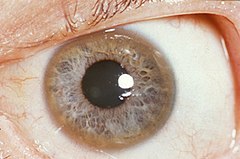
 Pomarańczowobrunatny pierścień Kaysera i Fleischera u 32-letniego pacjenta | |
| Klasyfikacje | |
| ICD-10 |
E83.0 |
|---|---|
| DiseasesDB | |
| OMIM | |
| MedlinePlus | |
| MeSH | |
Choroba Wilsona, zwyrodnienie soczewkowo-wątrobowe (ang. Wilson's disease, hepatolenticular degeneration) – uwarunkowane genetycznie, zaburzenie metabolizmu miedzi, prowadzące do osadzania się jej w nadmiernej ilości w tkankach organizmu. Miedź, która zwykle jest wydzielana z żółcią, gromadzi się początkowo w wątrobie, prowadząc do jej uszkodzenia. Po przekroczeniu możliwości magazynowania tego pierwiastka, trafia on z krwią do innych tkanek, duże koncentracje osiągając zwłaszcza w mózgu, rogówkach oczu i nerkach. Objawy neurologiczne choroby Wilsona wynikają z uszkodzenia jąder podstawnych mózgu.
Historia
Nazwa choroby upamiętnia brytyjskiego neurologa Samuela Alexandra Kinniera Wilsona, który opisał pierwsze cztery przypadki w 1912 roku. W 1948 roku J. N. Cumings wykazał, że wątroba i mózg chorych gromadzą duże ilości miedzi; cztery lata później Scheinberg i Gitlin dowiedli niedoboru ceruloplazminy w surowicy pacjentów. Penicylaminę do leczenia schorzenia wprowadził w 1956 roku Walsche. Defekt molekularny powodujący chorobę ostatecznie zidentyfikowały zespoły Tanziego i Bulla w 1993 roku.
Etiologia
Choroba Wilsona spowodowana jest dziedziczonymi autosomalnie recesywnie mutacjami genu ATP7B zlokalizowanego na chromosomie 13 (13q14.3), kodującego białko ATPazę ATP7B, którego funkcją jest aktywny transport miedzi w hepatocycie (wydalanie miedzi).
Stwierdzono ponad 200 różnych mutacji, z których te związane ze zmianami w domenie wiążącej ATP, powodują wcześniejsze i cięższe objawy kliniczne niż gdzie indziej zlokalizowane mutacje. Większość mutacji dotyczy wyłącznie kilku rodzin, a tylko kilka spotyka się częściej w populacjach.
Objawy
Pierwsze objawy pojawiają się zwykle między 10. a 40. rokiem życia. U prawie połowy chorych występują zaburzenia funkcji wątroby: zapalenie wątroby (przewlekłe lub ostre), marskość wątroby oraz nadostre zapalenie wątroby. U większości chorych pojawiają się zaburzenia neuropsychiatryczne, mogące prowadzić do nieprawidłowego rozpoznania (np. schizofrenii lub zespołu zależności alkoholowej). Zaburzenia psychiczne występują u około 65% chorych, w połowie przypadków wyprzedzając objawy neurologiczne. Najczęściej są to zaburzenia nastroju, zaburzenia osobowości, zaburzenia funkcji poznawczych, rzadziej objawy psychotyczne.
Jednymi z najczęstszych objawów są drżenia rąk, wybiórczy apetyt na potrawy, zaburzenia w pisaniu, ślinotok, zachwiania równowagi, wzmożone napięcie mięśni, zaburzenia mowy i połykania, ruchy mimowolne. Każde z tych czynników mogą na przestrzeni lat występować osobno i często powodują trudność w postawieniu diagnozy.
Charakterystycznym objawem choroby jest widoczny w rogówce oka pierścień Kaysera-Fleischera. Potwierdzenie jego obecności jest jedną z procedur diagnostycznych, innymi metodami są: badanie zawartości miedzi w wątrobie (próbkę uzyskuje się w wyniku biopsji) oraz badanie poziomu ceruloplazminy w surowicy. Jest to białko odpowiedzialne za magazynowanie miedzi; u większości chorych stwierdza się obniżenie jego zawartości.
Neuropatologia
Badanie makroskopowe: Poszerzenie komór bocznych, prążkowie i wzgórze są koloru brązowego lub rdzawego, w skorupie i/lub jądrze ogoniastym widuje się jamy.
Badanie mikroskopowe: utrata neuronów w skorupie, charakterystyczne są komórki Alzheimera typu 1 i typu 2, komórki Opalskiego.
Leczenie
Po ustaleniu rozpoznania rozpoczyna się leczenie farmakologiczne, które musi trwać do końca życia. Opiera się ono głównie na podawaniu leków usuwających z organizmu nadmiar miedzi i zapobiegających ponownemu jej gromadzeniu. Są to leki będące środkami chelatującymi, np. penicylamina lub trientyna, które zwiększają wydalanie miedzi. Stosowany jest także octan cynku, zmniejszający wchłanianie miedzi z przewodu pokarmowego. Wcześnie podjęte leczenie daje dobre rokowania, objawy zwykle znikają zupełnie. Pacjenci z ostrą niewydolnością wątroby spowodowaną tą chorobą są kwalifikowani do transplantacji tego narządu. Choroba wykryta, gdy powstały już nieodwracalne zmiany narządowe prowadzi do zgonu pacjenta. Zmniejszenie wchłaniania miedzi można uzyskać spożywając pokarmy ubogie w miedź, natomiast pokarmy bogate w miedź spożywając w połączeniu z białkami mleka.
Bibliografia
- Andrzej Szczeklik (red.): Choroby wewnętrzne: podręcznik multimedialny oparty na zasadach EBM. T. 1. Kraków: Medycyna Praktyczna, 2005, s. 936-939. ISBN 83-7430-031-0.
- Wojciech Kozubski, Paweł P Liberski, Maria Barcikowska: Choroby układu nerwowego. Warszawa: Wydaw. Lekarskie PZWL, 2004, s. 261-264. ISBN 83-200-2636-9.
- Anna Członkowska i inni, Wilson disease, „Nature Reviews. Disease Primers”, 4 (1), 2018, s. 21, DOI: 10.1038/s41572-018-0018-3, ISSN 2056-676X, PMID: 30190489, PMCID: PMC6416051 [dostęp 2021-06-29].
Linki zewnętrzne
- WILSON DISEASE w bazie Online Mendelian Inheritance in Man (ang.)