
| neuroblastoma | |
| Klasyfikacje | |
| ICD-10 | |
|---|---|

Nerwiak zarodkowy, nerwiak płodowy, zwojak zarodkowy (współczulny), nerwiak zarodkowy/płodowy współczulny, zwyczajowo neuroblastoma (łac. neuroblastoma) – złośliwy nowotwór wywodzący się z komórek cewy nerwowej (neuroblastów). Jest najczęstszym nowotworem rozpoznawanym u niemowląt. Blisko 50% jego przypadków występuje u dzieci poniżej 2. roku życia. W około 25–35% przypadków stwierdza się nieprawidłowości genetyczne pod postacią delecji w obrębie krótkiego ramienia chromosomu 1 (1p35-36). Obecność tej aberracji chromosomowej jest związana ze złym rokowaniem. Guz najczęściej rozwija się w rdzeniu nadnercza (40% przypadków), a rzadziej w przykręgosłupowych zwojach współczulnych w jamie brzusznej (25%), może się jednak rozwinąć w każdej części pnia współczulnego. Objawy nerwiaka płodowego są niezwykle różnorodne. Spowodowane są obecnością uciskającego tkanki guza, występowaniem przerzutów oraz produkcją katecholamin przez komórki nowotworu. Należą do nich, między innymi, niedokrwistość, zmniejszenie masy ciała, nadciśnienie tętnicze, zespół Hornera. W diagnostyce stosuje się biopsję aspiracyjną cienkoigłową, badania laboratoryjne (oznaczanie katecholamin w moczu), badania obrazowe (usg, rtg, scyntygrafia, tomografia komputerowa) i inne. Leczenie polega głównie na zabiegu chirurgicznym z ewentualną chemio- lub radioterapią. Rokowanie zależy od szeregu czynników, w tym od wieku dziecka, w którym wystąpił początek choroby. Nerwiak płodowy jest jednym z niewielu nowotworów złośliwych, które mogą ulec spontanicznej regresji, przechodząc od postaci niezróżnicowanej do łagodnej, dobrze zróżnicowanej.
Historia
Pierwszym, który opisał ten typ nowotworu, był Rudolf Virchow w 1864 roku, który określił go wtedy jako rodzaj glejaka. Guzy nowotworowe u dzieci przerzutujące do wątroby, płuc i kości opisywane były pod koniec XIX i na początku XX wieku jako choroba Peppera (William Pepper), guz Hutchisona (Robert Hutchison), guz Abercrombiego (John Abercrombie), zespół Parkera (R.W. Parker) i zespół Smitha (Jean Smith): wszystkie terminy mają dziś znaczenie historyczne (niekiedy używane są na określenie typowych przerzutów, o czym będzie mowa poniżej). W 1891 roku Marchand powiązał histologicznie nerwiaka płodowego ze zwojami układu współczulnego. W 1914 roku Karl Herxheimer wykazał, że włókna guza barwią się dodatnio na obecność znaczników neuronalnych. Dalszy postęp wiedzy o nerwiaku zarodkowym dokonał się w 1927 roku, gdy Harvey Cushing i S. Burt Wolbach pierwsi opisali transformację neuroblastoma do ganglioneuroma. Everson i Cole w 1966 roku spostrzegli, że ten typ przemiany nowotworu zachodzi rzadko u pacjentów starszych niż 6 miesięcy. Fenotyp 4S (S od specjalny) opisali D’Angio i wsp. w 1971 roku.
Epidemiologia
Nerwiak zarodkowy stanowi około 7% nowotworów wieku dziecięcego (do 15. roku życia) i odpowiada za około 15% zgonów dzieci z powodu nowotworów złośliwych. Jest najczęstszym pozaczaszkowym guzem litym wieku dziecięcego. Około 85% przypadków zachorowań ma miejsce w pierwszych 4 latach życia: ponad połowa chorych ma mniej niż 2 lata. Niektóre guzy są wrodzone. Neuroblastoma rzadko występuje po 10. roku życia. Opisywane są przypadki rodzinnego jej występowania; szacuje się, że rodzinny nerwiak zarodkowy stanowi 1-2% wszystkich przypadków guza. W Polsce notuje się 70–80 nowych zachorowań rocznie.
Częściej występuje u ludzi rasy białej niż u czarnej. Nieznacznie częściej (1,2 razy) neuroblastoma dotyka mężczyzn. Zaobserwowano też różnice geograficzne występowania nowotworu – w Stanach Zjednoczonych występuje z częstością 8–8,7:1 000 000 lub 9,5:1 000 000 dzieci, podczas gdy badania na populacji japońskiej w ramach szeroko prowadzonych badań przesiewowych (patrz niżej) w latach 1984–2002 pozwoliły oszacować częstość nowotworu na około 180:1 000 000.
Czynniki ryzyka
Stwierdzono większą częstość występowania nerwiaków płodowych w zespole Beckwitha-Wiedemanna i rzadkim zespole Simpsona-Golabiego-Behmela. Wykazywano statystycznie większe ryzyko zachorowania u dzieci matek:
- przyjmujących fenytoinę w czasie ciąży (ryzyko zwiększone w niewielkim stopniu)
- często spożywających alkohol w czasie ciąży
- używających koloryzujących farb do włosów
- stosujących hormony płciowe
- leczących się diuretykami.
Nie stwierdzono związku neuroblastomy z paleniem papierosów przez matkę, spożyciem kawy i ekspozycją na medyczne źródła promieniowania jonizującego.
Patogeneza
Nerwiak zwojowy rozwija się z wielokierunkowych prekursorowych komórek cewy nerwowej różnicujących się w kierunku neuroblastów, komórek zwojowych i komórek Schwanna. Zaproponowano, że komórki Schwanna spotykane w tkance guza mogą wydzielać czynniki indukujące jego wzrost.
Nerwiak węchowy zarodkowy (esthesioneuroblastoma, ang. olfactory neuroblastoma) jest bardzo rzadkim guzem podobnym do neuroblastomy histologicznie, ale o odmiennej biologii, wywodzącym się z nabłonka węchowego.
Genetyka
W około 25-35% przypadków nerwiaka zarodkowego stwierdza się nieprawidłowości genetyczne pod postacią delecji w obrębie krótkiego ramienia chromosomu 1 (1p35-36). Obecność tej delecji jest związana ze złym rokowaniem.
Niekorzystnym czynnikiem prognostycznym jest również amplifikacja, czyli zwielokrotnienie liczby kopii protoonkogenu N-myc (MYCN) nawet do kilkuset. Protoonkogen normalnie znajduje się na krótkim ramieniu chromosomu 2, podczas gdy w komórkach neuroblastomy stwierdza się tzw. podwójne malutkie chromosomy (ang. double minutes) – bardzo małe fragmenty chromosomów – i regiony jednolicie barwiące się (ang. homogenous staining regions- HSR) w chromosomach 2, 4, 9, 12 – regiony z licznymi kopiami onkogenu. Amplifikacja MYCN występuje w około 20% guzów, a im więcej kopii onkogenu, tym gorsze rokowanie; prognostyk ten został uwzględniony w najnowszej klasyfikacji nerwiaków płodowych (International Neuroblastoma Pathology Classification).
Lokalizacja i obraz makroskopowy
Guz najczęściej rozwija się w obrębie jamy brzusznej (65%), z czego przynajmniej połowa ma punkt wyjścia w rdzeniu nadnercza. Przestrzeń zaotrzewnowa stanowi miejsce wyjścia łącznie około 80% wszystkich guzów. Inne częste lokalizacje to przykręgosłupowe zwoje współczulne w śródpiersiu tylnym (15%), miednicy (2-5%), szyi (2-5%), wyjątkowo mózgowie. Neuroblastoma przybiera różne rozmiary, może być mikroskopową zmianą lub guzem o masie przekraczającej 1000 g. Guz jest miękki, ma konsystencję mózgu, jest kruchy, z ogniskami krwotoków, martwicy, zwapnień; występują w nim torbiele.
W momencie rozpoznania choroba jest zlokalizowana (bez przerzutów) w 40% przypadków. U około połowy pacjentów można stwierdzić szerzenie się przerzutów drogą krwionośną.
Guz nacieka okoliczne tkanki i daje przerzuty do:
- węzłów chłonnych zaotrzewnowych i innych
- wątroby (przerzut typu Peppera)
- kości, szczególnie czaszki, oczodołów (typ Hutchinsona)
- skóry (typ Smitha)
- szpiku kostnego
- płuc
- mózgu.
Neuroblastoma in situ

Komórki guza stwierdzano również w badaniach autopsyjnych w korze nadnerczy martwo urodzonych płodów lub zmarłych w okresie okołoporodowym wcześniaków i donoszonych noworodków (neuroblastoma in situ).
Objawy
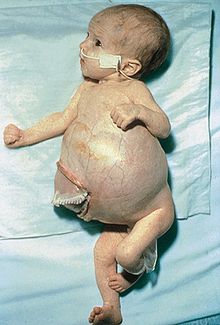


Objawy w nerwiaku płodowym są niezwykle różnorodne. Spowodowane są obecnością uciskającego tkanki guza, występowaniem przerzutów oraz produkcją katecholamin przez komórki nowotworu.
- Nieswoiste objawy spowodowane przerzutami
- utrata masy ciała
- podwyższona temperatura
- senność
- bladość
- osłabienie
- drażliwość, niepokój
Objawy spowodowane przez ucisk guza lub obecność przerzutów w zależności od lokalizacji:
- Szyja
- macalny guz
- zespół Hornera w przypadku umiejscowienia w zwojach szyjnych pnia współczulnego
- Oczodół i gałka oczna
- wybroczyny i podbiegnięcia krwotoczne w okolicy oczodołu (krwiaki okularowe, „oczy szopa pracza”, ang. raccoon eyes), wytrzeszcz gałki ocznej
- obrzęk powiek i spojówek
- obrzęk tarczy nerwu wzrokowego
- zanik nerwu wzrokowego
- krwawienia do siatkówki
- zez
- Klatka piersiowa (śródpiersie tylne)
- duszność, kaszel, stridor
- infekcje płuc
- ból w klatce piersiowej
- zaburzenia połykania
- obrzęk szyi, zespół żyły głównej górnej (lokalizacja w śródpiersiu tylnym)
- bez objawów (guzy umiejscowione w dolnej części klatki piersiowej)
- Jama brzuszna
- rosnący, macalny guz, przemieszczający nerkę przednio-bocznie i do dołu
- brak łaknienia
- wymioty
- ból brzucha
- zmniejszenie masy ciała
- nadciśnienie tętnicze spowodowane uciskiem naczyń nerkowych przez guz
- hepatomegalia w przypadku przerzutów do wątroby
- gwałtowny wzrost guza, ból, bladość powłok, hipotonia tętnicza w przypadku krwotoku do guza
- Okolica przykręgosłupowa
- zlokalizowany ból pleców
- zaburzenia neurologiczne spowodowane uciskiem korzeni nerwów grzbietowych
- paraplegia
- zespół ogona końskiego
- ataksja móżdżkowa
- przeczulica
- zaniki mięśniowe
- skolioza
- zaburzenia funkcji pęcherza moczowego
- zaburzenia funkcji zwieracza odbytu
- Miednica
- zaparcia
- niepokój w czasie oddawania moczu
- zastój moczu
- masa guza wyczuwalna w badaniu per rectum
- Kościec
- niedokrwistość z powodu niewydolności szpiku
- bóle kości
- niepokój (u młodszych dzieci)
- Skóra i tkanka podskórna
- mnogie guzki podskórne (charakterystyczne dla IV fazy neuroblastomy u niemowląt, rzadko poza okresem wczesnego dzieciństwa). Mają postać licznych wykwitów grudkowych lub guzkowych średnicy 2–20 mm, o niebieskawym kolorze (określane niekiedy w piśmiennictwie anglojęzycznym jako blueberry muffins – bułeczki nadziewane jagodami). Wykwity mają tendencję do blednięcia w części środkowej i tworzenia się rumieniowatej otoczki po 2–3 minutach od podrażnienia zmiany przez potarcie; objawy te są związane z uwalnianiem obkurczających naczynia krwionośne katecholamin przez komórki guza. Zmiany skórne mogą występować na powierzchni całego ciała, najczęściej w obrębie tułowia i kończyn.
Zespoły paraneoplastyczne
- Objawy ogólne spowodowane nadmierną produkcją katecholamin
- nadciśnienie tętnicze
- napadowe zaczerwienienie twarzy, pocenie się
- kołatanie serca.
Objawy takie opisywano u ciężarnych z wrodzonym nerwiakiem płodu („mirror syndrome”); w jednym przypadku guz przerzutował do łożyska.
- Objawy ogólne spowodowane nadmierną produkcją VIP
- wodniste biegunki
- bóle brzucha
- atonia jelit
- nasilona hipokalemia
- Encefalopatia móżdżkowa (zespół mioklonii i opsoklonii)
Dotyczy około 2-4% pacjentów z neuroblastomą; jego etiologia nie jest znana. Składają się na niego gwałtowne, chaotyczne ruchy gałek ocznych, postępująca ataksja i nieregularne, miokloniczne skurcze mięśni. Objawy zazwyczaj ustępują po usunięciu ogniska pierwotnego, ale u 70-80% pacjentów pozostają długotrwałe deficyty neurologiczne.
Obraz histologiczny



Histologicznie, w zależności od stopnia dojrzałości komórek, rozróżniamy:
- nerwiaka płodowego – neuroblastomę
- nerwiaka płodowego zwojowego – ganglioneuroblastomę.
Neuroblastoma należy do tzw. guzów drobnookrągłoniebieskokomórkowych: charakterystyczne dla obrazu histologicznego są skupiska małych (nieco większych od limfocytów) prymitywnych komórek z okrągłymi lub owalnymi hiperchromatycznymi jądrami i małą ilością cytoplazmy. Ziarnista chromatyna przyrównywana jest do ziaren soli i pieprzu (ang. salt and pepper chromatin). Widoczne są liczne figury podziałów mitotycznych, komórki apoptotyczne i tak zwane rozetki Homera Wrighta, złożone z komórek nowotworowych wianuszkowato otaczających bladoróżowy fibrylarny materiał utworzony z ich cienkich wypustek cytoplazmatycznych.
W badaniu immunohistochemicznym stwierdza się dodatnią reakcję komórek nowotworu na obecność synaptofizyny, chromograniny, neurofilamentów i neuronospecyficznej enolazy (NSE); ujemna jest reakcja na wimentynę, desminę i keratynę.
W oparciu o obraz histologiczny guza opracowano kilka klasyfikacji histologicznych, cennych przy określaniu rokowania nowotworu.
Klasyfikacja Shimady i wsp. (1984)
Klasyfikacja Shimady opiera się na czterech kryteriach. Są to:
- charakter podścieliska: nowotwory bogatopodścieliskowe zawierają dużą ilość podścieliska zbudowanego z wrzecionowatych komórek typu komórek Schwanna (S100+) i przypominają pod tym względem takie nowotwory jak neurofibroma lub schwannoma. Nowotwory ubogopodścieliskowe zawierają z kolei podścielisko drobnowłókienkowe typu neuropile;
- odsetek komórek różnicujących się w kierunku komórek zwojowych: typ dojrzewający zawiera >5% neuroblastów dojrzewających do komórek zwojowych, typ niedojrzały (niezróżnicowany) zawiera mniej niż 5% takich komórek;
- indeks mitotyczno-kariokinetyczny (MKI, ang. mitosis-karyorrhexis index): oznacza on liczbę komórek dzielących się, piknotycznych i apoptotycznych z cechami karyorrhexis oznaczoną na 5000 komórek nowotworu. MKI niski oznacza liczbę takich komórek mniejszą niż 100; MKI średni wynosi mniej niż 200; MKI wysoki wynosi więcej niż 200;
- wiek pacjenta: klasyfikacja Shimady dzieli pacjentów na trzy grupy wiekowe, poniżej 18 miesięcy, od 18 miesięcy do 5 lat i powyżej 5 lat.
| Typ | Wiek | Korzystna charakterystyka histologiczna | Niekorzystna charakterystyka histologiczna |
|---|---|---|---|
| Bogatopodścieliskowy | Dobrze zróżnicowany Mieszany |
Guzkowy | |
| Ubogopodścieliskowy | |||
| < 18 miesięcy | MKI < 200 | MKI > 200 | |
| 18-60 miesięcy | MKI < 100 i typ dojrzewający | MKI > 100 lub niedojrzały | |
| > 5 lat | Żadne | Wszystkie |
Klasyfikacja Joshiego i wsp. (1992)
Klasyfikacja Joshiego nie ocenia guzów typu ganglioneuroblastoma (czyli takich, w których ponad 50% utkania stanowi dojrzałe utkanie ganglioneuronalne). Wyróżnia trzy stopnie zróżnicowania nerwiaka płodowego:
| Stopień | Liczba mitoz/DPW i obecność zwapnień | Przeżycie 5-letnie |
|---|---|---|
| I | <10 mitoz i zwapnienia | 89% |
| II | <10 mitoz lub zwapnienia | 77% |
| III | >10 mitoz bez zwapnień | 33% |
(DPW- duże pole widzenia mikroskopu)
| Stopień | Wiek | Amplifikacja MYCN | Typ według Shimady | Ploidia DNA | Rokowanie |
|---|---|---|---|---|---|
| 1 | 0-21 |
±
|
Każdy | Każda | Dobre |
| 2A/2B | <1 |
±
|
Każdy | Każda | Dobre |
| ≥1-21 |
-
|
Każdy |
-
|
Dobre | |
| ≥1-21 |
+
|
Korzystny |
-
|
Złe | |
| ≥1-21 |
+
|
Niekorzystny |
-
|
Złe | |
| 3 | <1 |
-
|
Każdy | Każda | Średnie |
| <1 |
+
|
Każdy | Każda | Złe | |
| ≥1-21 |
-
|
Korzystny |
-
|
Średnie | |
| ≥1-21 |
-
|
Niekorzystny |
-
|
Złe | |
| ≥1-21 |
+
|
Złe |
-
|
Złe | |
| 4 | <1 |
-
|
Każdy | Każda | Średnie |
| <1 |
+
|
Każdy | Każda | Złe | |
| ≥1-21 |
±
|
Każdy |
-
|
Złe | |
| 4S | <1 |
-
|
Korzystny | >1 | Dobre |
| <1 |
-
|
Każdy | =1 | Średnie | |
| <1 |
-
|
Niekorzystny | Każda | Średnie | |
| <1 |
+
|
Każdy | Każda | Złe |
Diagnostyka



Klasyfikacja oceny zaawansowania choroby pn. International Neuroblastoma Staging System (INSS) wymaga do rozpoznania: dodatniego wyniku biopsji szpiku i podwyższonego stężenia metabolitów katecholamin w dobowej zbiórce moczu albo dodatniego wyniku biopsji guza. Natomiast u noworodków wymagany jest wzrost stężenia katecholamin i dodatni wynik scyntygrafii MIGB (po 14 dniu życia). Przed 90 dniem życia nie jest wymagana biopsja szpiku.
Rozpoznanie stawiane jest na podstawie histopatologicznego potwierdzenia choroby w bioptacie guza lub biopsji szpiku i podwyższonego poziomu katecholamin w osoczu lub ich metabolitów w moczu.
Obligatoryjne do oceny stopnia zaawansowania są: biopsja szpiku, LDH, NSE, Ferrytyna, scyntygrafia kości.
Wkłucie lędźwiowe nie jest zalecane jeśli istnieje podejrzenie nerwiaka zarodkowego ponieważ może powodować rozsianie choroby.
Ocenie wielkości ogniska pierwotnego i ewentualnych przerzutów służą badania obrazowe:
- rtg jamy brzusznej i klatki piersiowej
- scyntygrafia celem oceny ogniska pierwotnego z użyciem 123I-mIBG (znakowanej jodem-123 benzyloguanidyny) (zamiast jodem -131)
- scyntygrafia kośćca z użyciem izotopu technetu celem obrazowania przerzutów
- tomografia komputerowa lub MRI okolicy guza
- mielografia w przypadku objawów neurologicznych.
Stopnie zaawansowania
Przyjęty system oceny klinicznego zaawansowania nowotworu Evansa dzieli pacjentów na pięć grup.
| Stadium | Opis |
|---|---|
| I | Guz ograniczony do jednego narządu, możliwy do usunięcia w całości. |
| II | Guz nacieka poza narząd, w którym się rozwinął, nie przekracza jednak linii środkowej ciała. Węzły chłonne po stronie, w której rozwija się nowotwór zajęte lub nie. |
| III | Guz przekracza linię środkową ciała, ipsilateralne węzły chłonne z przerzutami lub bez. |
| IV | Odległe przerzuty drogą krwi do narządów wewnętrznych: płuc, wątroby, mózgu, do tkanek miękkich i odległych węzłów chłonnych. |
| IVS | Niemowlęta z małym guzem nadnercza (jak w stadium I i II), przerzutami do wątroby, szpiku kostnego lub skóry, bez zniszczenia struktury kości. |
Mimo że powszechnie używa się systemu INSS, trwają prace nad nowymi wytycznymi. W 2005 roku onkolodzy dziecięcy z International Neuroblastoma Risk Group (INRG) zaproponowali, na podstawie 8800 przypadków neuroblastomy z Europy, Japonii, Kanady, Stanów Zjednoczonych i Australii z lat 1990–2002, nowy system klasyfikacji guzów. Badania retrospektywne wykazały wysoki współczynnik przeżyć w grupie wiekowej 12–18 miesięcy, wcześniej kategoryzowanej jako grupa wysokiego ryzyka, co uzasadniło decyzję o przeklasyfikowaniu pacjentów w wieku 12–18 miesięcy bez amplifikacji MYCN do grupy średniego ryzyka.
Nowa klasyfikacja guzów w momencie rozpoznania według International Neuroblastoma Risk Group (International Neuroblastoma Risk Group Staging System, INRGSS):
- Stopień L1: zlokalizowana choroba bez czynników ryzyka
- Stopień L2: zlokalizowana choroba z czynnikami ryzyka
- Stopień M: rozsiana choroba
- Stopień MS: rozsiana choroba, typ „specjalny” (odpowiada 4S).
Leczenie
- Stopień I zaawansowania
Wystarczające jest chirurgiczne całkowite usunięcie zmiany.
- Stopień II zaawansowania
Zaleca się krótkotrwałą chemioterapię w wypadku zajęcia węzłów chłonnych. W przypadku penetracji guza do kanału kręgowego nie zaleca się rutynowo laminektomii i napromieniania, ale również tylko chemioterapię.
- Stopień III i IV zaawansowania
Stosuje się agresywne, skojarzone leczenie chemiczne, chirurgiczne i radioterapię według bieżącego protokołu (ESIOP-HRNBL). Protokół składa się z chemioterapii indukcyjnej (cyklofosfamid, winkrystyna, etopozyd, cisplatyna, karboplatyna), w przerwach między blokami podaje się także G-CSF (filgrastim). Po zakończeniu chemioterapii pobiera się komórki macierzyste do późniejszej megachemioterapii, a następnie podejmuje się próbę resekcji całkowitej guza pierwotnego. Kolejnym etapem leczenia jest zastosowanie megachemioterapii z autogenicznym przeszczepieniem komórek macierzystych. Miejsce, w którym znajdował się guz pierwotny poddaje się napromienianiu. Po zakończeniu radioterapii, przeprowadza się terapię choroby resztkowej izotreotyniną albo monoklonalnymi przeciwciałami anty-GD2.
- Stopień IVS zaawansowania
W zależności od oceny czynników ryzyka (skala Philadelphia) na podstawie stanu ogólnego pacjenta można w ogóle nie prowadzić leczenia i obserwować pacjenta (watch and wait) lub prowadzi się krótkotrwałą chemioterapię.
Terapie eksperymentalne

Inhibitory topoizomerazy 1
Topotekan i irynotekan są mało toksyczne i dają dobre efekty, zwłaszcza w połączeniu z cyklofosfamidem.
Radioterapia celowana
Immunoterapia
GD2 jest disjalogangliozydem ulegającym wysokiej ekspresji w komórkach guza; celowane terapie anty-GD2 są w I i II fazie badań klinicznych.
Retinoidy
Randomizowane badanie nad skutecznością kwasu 13-cis-retinowego po chemioterapii ablacyjnej pozwoliło wykazać potencjalną skuteczność retinoidów w terapii pacjentów z grupy wysokiego ryzyka.
Inhibitory angiogenezy
Unaczynienie guza koreluje z agresywnością fenotypu; inhibitory angiogenezy stanowią atrakcyjną opcję terapeutyczną. Badania przedkliniczne dały jak dotąd rozbieżne wyniki.
Inhibitory kinaz tyrozynowych
Małocząsteczkowy inhibitor kinazy Trk, CEP-701 (KT-6587), wykazywał wysoką skuteczność hamowania wzrostu komórek neuroblastomy in vivo. Trwają badania kliniczne I fazy nad tym lekiem.
Inhibitory deacetylazy histonów
Aktualne i zakończone badania kliniczne nad nowymi terapiami
Ostatnie badania skupiają się na ograniczeniu działań terapeutycznych w grupie pacjentów niskiego i pośredniego ryzyka, przy utrzymaniu przeżywalności rzędu 90%. Badanie na 467 pacjentów niskiego ryzyka (A3961) prowadzone od 1997 do 2005 potwierdziło hipotezę, że leczenie w tej podgrupie chorych może być zredukowane. Pacjenci z korzystnymi czynnikami rokowniczymi (grading guza i odpowiedź na leczenie) otrzymali cztery cykle chemioterapii, a ci z niekorzystną charakterystyką choroby osiem cykli, z trzyletnim całkowitym przeżyciem i ogólną przeżywalnością rzędu 90% dla całej kohorty badanych. Planuje się zintensyfikowanie leczenia u pacjentów z aberracjami chromosomowymi 1p36 lub 11q23, jak również u tych chorych, którzy nie odpowiadają na leczenie.
Z drugiej strony, w ostatnich 15 latach intensyfikowano leczenie w grupie pacjentów wysokiego ryzyka. Sprawdzano skuteczność różnych protokołów indukcji chemioterapii, zmiany czasu zabiegu chirurgicznego, przeszczepów komórek pnia, zmian dawkowania radioterapii, a także przeciwciał monoklonalnych i retinoidów w zwalczaniu choroby resztkowej. Ostatnie badania kliniczne III fazy mające na celu zwiększenie przeżywalności pacjentów grupy wysokiego ryzyka pozwoliły (lub pozwolą) odpowiedzieć na następujące pytania:
- 1991–1996: Pierwsze badanie z randomizacją (faza III) na grupie 379 pacjentów wysokiego ryzyka przeprowadzone przez Children’s Cancer Group (CCG-3891) dowiodło zwiększonego przeżycia po zastosowaniu terapii mieloablacyjnej (drogą napromieniania całego ciała) i kwasu 13-cis-retinowego (Accutane)
- 1996–2003: Niemieckie (GPOH) badanie z randomizacją NB97 miało na celu porównanie przebiegu choroby u 295 pacjentów wysokiego ryzyka poddanych przeszczepowi komórek pnia lub skonsolidowanej chemioterapii. Wyniki dowodziły wyższej przeżywalności w podgrupie której przeszczepiono komórki pnia
- 2000–2006: Badanie COG-A3973 na grupie 486 pacjentów miało odpowiedzieć na pytanie kliniczne odnośnie do zastosowania oczyszczonych komórek pnia w protokole CEM-LI (karboplatyna, etopozyd, melfalan, miejscowe napromienianie). Przeszczepione komórki pnia nie zwiększyły przeżycia pacjentów
- 2000–2012: Kolejne badanie (COG-ANBL0032) na 423 pacjentach ma wykazać, czy przeciwciało ch14.18 z IL-2 i GMCSF (niemieckie badania retrospektywne GPOH NB90 i NB 97 z małymi dawkami i bez leczenia cytokinami) zwiększy przeżywalność chorych.
- 2002–2008: Międzynarodowe Towarzystwo Onkologii Dziecięcej (SIOP, International Society of Paediatric Oncology) utworzyło europejską grupę na rzecz walki z nerwiakiem zarodkowym (European SIOP Neuroblastoma Group, ESIOP NB) w 1994 roku, a badania kliniczne protokołu dla neuroblastomy wysokiego ryzyka weszły w III fazę w 2002 roku (SIOP-EUROPE-HR-NBL-1). Zastosowano „przyspieszony” protokół COJEC (8 cykli chemioterapii oddzielonych dziesięciodniowymi przerwami), po którym przeszczepiono komórki pnia i w jednej grupie zastosowano CEM (karboplatynę, etopozyd, melfalan), a w drugiej BuMel (busulfan, melfalan) i następnie w jednej grupie wdrożono leczenie przeciwciałami ch14.18, a w drugiej nie. Badanie pozwoli ocenić skuteczność czynników wzrostu i porównać protokoły przeszczepów z lub bez zastosowania przeciwciał ch14.18l wszyscy pacjenci otrzymają kwas 13-cis-retinowy. W badaniu weźmie udział 1000 pacjentów (175 na rok).
- 2005–2010: Trwające niemieckie badanie z randomizacją NB2004 na grupie 642 pacjentów wszystkich grup ryzyka (około połowa wysokiego ryzyka) ma na celu sprawdzenie skuteczności topotekanu w połączeniu z terapią MIBG. Protokół dla grupy wysokiego ryzyka przewiduje sześciomiesięczne leczenie kwasem 13-cis-retinowym po przeszczepie, następnie 3 miesiące przerwy i kolejne trzy miesiące podawania kwasu 13-cis-retinowego.
- 2007: Badanie kliniczne III fazy COG (ANBL0532) zainaugurowane w grudniu 2007 na grupie 495 pacjentów ma na celu porównanie pojedynczych i podwójnych przeszczepów; indukcja chemioterapii przewiduje dwa cykle topotekanu
Oprócz tych badań w III fazie, instytucje badawcze takie jak Baylor College of Medicine/Texas Children’s, St. Jude Children’s Research Hospital (Memphis, Tennessee), czy Memorial Sloan-Kettering Cancer Center w Nowym Jorku oferują własne, odmienne protokoły leczenia. Texas Children’s wprowadziło metodę indukcji chemioterapii („chemo-switching”) polegającą na podawaniu pulsów dużych dawek cisplatyny i małych dawek etopozydu przez kilka tygodni pierwszych dwóch cykli chemioterapii. Szpital St Jude’s bada obecnie nowy protokół, zawierający irinotecan i gefitinib z 16 miesiącami chemioterapii podtrzymującej po przeszczepie komórek pnia (naprzemiennie kwas 13-cis-retinowy i topotekan). Centrum Sloan-Kettering oferuje leczenie przeciwciałami monoklonalnymi 3F8, wprowadzonymi do leczenia w połowie lat 80. Przeciwciała te stosowane są w leczeniu choroby resztkowej lub w fazie konsolidacji chemioterapii zamiast przeszczepu komórek pnia.
Spontaniczna regresja
Opisano niezwykłe zjawisko spontanicznej (samoistnej), całkowitej regresji guza w stadium rozsianym. Zjawisko to obserwowano wyłącznie w nerwiakach niemowlęcych (wykrywanych u dzieci przed ukończeniem pierwszego roku życia). Typ określany jako 4S stanowi około 5% przypadków neuroblastomy.
Rokowanie i czynniki prognostyczne
Ogólna przeżywalność wynosi 55% (prawie 100% w stopniu I, 75% w II, 43% w III, 15% w IV, 70–80% w IVS).
Czynniki rokownicze można podzielić na trzy główne grupy:
- czynniki rokownicze kliniczne
- histologiczne
- biologiczne.
- Czynniki kliniczne rokowania
- wiek dziecka (niemowlęta mają bardzo dobre rokowanie, a dzieci po 2 roku życia znacznie gorsze)
- umiejscowienie ogniska pierwotnego (jama brzuszna jest lokalizacją niekorzystną, guz szyi, śródpiersia, miednicy rokuje lepiej)
- wczesna normalizacja poziomu katecholamin w surowicy po rozpoczęciu leczenia (do miesiąca)
- wysoki poziom ferrytyny: > 142 μg/dl (niekorzystny)
- mnogie przerzuty w kościach (niekorzystne)
- wysoki poziom LDH w surowicy >1500 j. U. (niekorzystny)
- stosunek osoczowego poziomu kwasu wanilinomigdałowego do homowanilinowego (mniejszy niż 1 wiąże się z gorszym rokowaniem)
- stężenie neuronospecyficznej enolazy w surowicy (NSE) >100 ng/ml rokuje źle
- Czynniki histologiczne rokowania
- charakterystyka histologiczna według Shimady (zob. wcześniej)
- charakterystyka histologiczna według Joshiego – obecność zwapnień (zob. wcześniej)
- Czynniki biologiczne i genetyczne rokowania
- obecność delecji 1p35-36 (związana ze złym rokowaniem)
- inne delecje: 11q, 14q, 17q
- naddatek 17q (ang. 17q gain, niekorzystny), także na chromosomach 6, 7, 18
- amplifikacja MYCN >10 kopii jest uznawana za najbardziej niekorzystny czynnik rokowania
- koamplifikacja DDX1
- ploidia guza (guzy okołodiploidalne mają gorsze, hiperdiploidalne i okołotriploidalne lepsze rokowanie)
- ekspresja CD 44 (jest korzystna rokowniczo)
- wysoki poziom ekspresji nerwowego czynnika wzrostu TrkA wiąże się z lepszym rokowaniem.
Częste są wznowy nowotworu: w takim przypadku wymagane jest ponowne leczenie. Może to stanowić problem, ponieważ niektóre metody terapeutyczne, takie jak chemioterapia, mają ograniczoną skuteczność, wynikającą z tego, że źródłem nawrotu są komórki najbardziej oporne na cytostatyki, które były w stanie przetrwać początkowe leczenie.
Intensywna chemioterapia i radioterapia dają późne negatywne skutki. Oszacowano, że u dwóch osób z trzech, które przeżyją nowotwór w dzieciństwie, rozwinie się przynajmniej jedna przewlekła choroba, a czasami zagrażający życiu stan zdrowotny, w ciągu 20–30 lat od postawienia diagnozy nowotworu.
Prewencja i badania przesiewowe
Podjęto próby badań przesiewowych neuroblastomy metodami wykrywającymi metabolity katecholamin w próbkach moczu; programy badań przesiewowych prowadzone od lat 1980. obejmowały trzytygodniowe noworodki w Japonii, trzytygodniowe noworodki i sześciomiesięczne niemowlęta w Kanadzie i roczne dzieci w Niemczech.
Japonia rozpoczęła badania przesiewowe w kierunku nerwiaka płodowego u sześciomiesięcznych niemowląt poprzez badanie stężenia kwasu homowanilinowego (HVA) i kwasu wanilinomigdałowego (VMA) w moczu w 1984. Badania przesiewowe zostały wstrzymane w 2004 po ukazaniu się wyników badań przeprowadzonych w Kanadzie i Niemczech, z których wynikało, że prowadzenie badań przesiewowych nie zmniejsza ryzyka zgonu z powodu nerwiaka płodowego, ale stwarza ryzyko postawienia diagnozy nowotworu, który może samoistnie ulec regresji i niepotrzebnie wystawia dzieci na zabiegi chirurgiczne i chemioterapię.
Duże badania kanadyjskie wykazały, że kobiety w ciąży, które brały zestawy witamin wzbogacone o kwas foliowy przed i w trakcie 3 pierwszych miesięcy ciąży, miały obniżone ryzyko wystąpienia neuroblastomy u ich dzieci o 60%.
Bibliografia
- V. Kumar, R. Cotrani, S. Robbins: Patologia Robbinsa. Urban&Partner, 2005. ISBN 978-83-89581-92-1.
- Jerzy Stachura, Wenancjusz Domagała: Patologia znaczy słowo o chorobie. Kraków: Wydawnictwo PAU, 2003. ISBN 83-88857-65-7.
- Onkologia dziecięca. W: Dorota Perek: Pediatria. Podręcznik do Państwowego Egzaminu Lekarskiego i egzaminu specjalizacyjnego. Anna Dobrzańska, Józef Ryżko (red.). Wrocław: Urban&Partner, 2005, s. 621-625. ISBN 83-89581-25-6.
- Choroby nowotworowe u dzieci. W: Urszula Radwańska: Pediatria. Krystyna Kubicka, Wanda Kawalec (red.). Warszawa: 2003, s. 459-461. ISBN 83-200-2773-X.
- Maris J.M., Hogarty M.D., Bagatell R., Cohn S.L.. Neuroblastoma. „Lancet”. 369. 9579, s. 2106-20, 2007. DOI: 10.1016/S0140-6736(07)60983-0. PMID: 17586306.
- Howman-Giles R., Shaw P.J., Uren R.F., Chung D.K.. Neuroblastoma and other neuroendocrine tumors. „Semin Nucl Med”. 37. 4, s. 286-302, 2007. PMID: 17544628.
- Gael J. Lonergan, Cornelia M. Schwab, Eric S. Suarez, Christian L. Carlson. Neuroblastoma, Ganglioneuroblastoma, and Ganglioneuroma: Radiologic-Pathologic Correlation. „Radiographics”. 22. 4, s. 913-934, 2002.
- Josée Brossard, Mark L. Bernstein, Bernard Lemieux: Neuroblastoma: an enigmatic disease. British Medical Bulletin 52:787-801 (1996)
- Acharya S., Jayabose S., Kogan S.J., et al. Prenatally diagnosed neuroblastoma. „Cancer”. 80. 2, s. 304-10, 1997.
- Castleberry RP. Predicting outcome in neuroblastoma. „New England Journal of Medicine”. 340. 25, s. 1992-3, 1999.
- Brian H. Kushner. Neuroblastoma: A Disease Requiring a Multitude of Imaging Studies. „The Journal of Nuclear Medicine”. 45. 7, s. 1172-1188, 2004.
- Elżbieta Adamkiewicz-Drożyńska. Czynniki prognostyczne i nowe możliwości leczenia neuroblastoma. „Współcz Onkol”. 4. 2, s. 72-75, 2000.
- Weinstein J.L., Katzenstein H.M., Cohn S.L.. Advances in the diagnosis and treatment of neuroblastoma. „Oncologist”. 8. 3, s. 278-92, 2003. PMID: 12773750.
- Andrzej I. Prokurat: Rozdział 78. W: Maciej Bagłaj, Piotr Kaliciński: Chirurgia dziecięca. Warszawa: PZWL, 2016. ISBN 978-83-200-5014-1.
- Rozdział 6.2. W: Andrzej Chilarski: Zarys zagadnień chirurgii wieku rozwojowego. Warszawa: PTCHD, 2009. ISBN 978-83-61997-04-7.
Linki zewnętrzne
- Byron D Joyner: Neuroblastoma. eMedicine.
- Neuroblastoma na stronie American Cancer Society (ang.)
- Neuroblastoma Society (ang.)
- NEUROBLASTOMA w bazie Online Mendelian Inheritance in Man (ang.)