
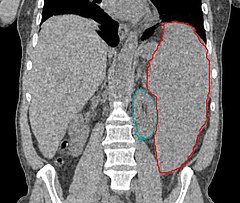
 Znacznie powiększona śledziona (zaznaczona na czerwono, na zielono zaznaczono nerkę) w tomografii komputerowej | |
| Klasyfikacje | |
| ICD-10 |
D46 |
|---|---|
| D46.0 |
Oporna niedokrwistość bez syderoblastów |
| D46.1 |
Oporna niedokrwistość z syderoblastami |
| D46.2 |
Oporna niedokrwistość z nadmiarem blastów |
| D46.3 |
Oporna niedokrwistość z nadmiarem blastów z transformacją |
| D46.4 |
Oporna niedokrwistość, nieokreślona |
| D46.7 |
Inne zespoły mielodysplastyczne |
| D46.9 |
Zespół mielodysplastyczny, nieokreślony |
| ICDO | |
| DiseasesDB | |
| MeSH | |
Zespoły mielodysplastyczne (mielodysplazja szpiku) (ang. myelodysplastic syndrome, MDS) – klonalne zaburzenie hematopoetycznej komórki macierzystej uniemożliwiające różnicowanie i dojrzewanie komórek, przejawiające się jedno-, dwu- lub trójliniową cytopenią i najczęściej bogatokomórkowym szpikiem. Jest to grupa blisko związanych jednostek chorobowych, w których proces tworzenia krwinek jest zakłócony przez niezdolność niedojrzałych komórek do prawidłowego wzrostu i rozwoju. Czasami nazywany stanem przedbiałaczkowym, ponieważ w zaawansowanych postaciach stosunkowo często transformuje w kierunku ostrych białaczek.
Epidemiologia
Na zespoły mielodysplastyczne chorują częściej mężczyźni w wieku 60–75 lat. Zapadalność w Europie szacuje się na 2,1-12,6/100 000, przy czym powyżej 70. roku życia wzrasta do 15-50/100 000.
Etiopatogeneza
Przyczyny powstawania zespołu mielodysplastycznego nie są do końca poznane. Postuluje się udział szeregu czynników ryzyka, które mogą zwiększać częstotliwość zachorowania na MDS.
Czynniki ryzyka
Do czynników ryzyka mogą należeć:
- narażenie na związki chemiczne takie jak: pestycydy, toluen, benzen, środki ochrony roślin, nawozy sztuczne, farby do włosów, ksylen, chloramfenikol
- dym tytoniowy
- metale ciężkie
- promieniowanie jonizujące
- leki – cytostatyki
- niedokrwistość aplastyczna
- czynniki genetyczne – większa częstość MDS u ludzi z niedokrwistością Fanconiego, chorobą von Recklinghausena i zespołem Downa.
Patogeneza
Istotą zespołów mielodysplastycznych jest dysproporcja pomiędzy normo- lub nawet bogatokomórkowym szpikiem a występującą w krwi obwodowej cytopenią (zmniejszeniem ilości składników morfotycznych krwi). Jest to związane z zaburzeniem proliferacji, dojrzewania i czasu przeżycia powstających komórek. W zależności od rodzaju zespołu, dominują różne zaburzenia hemopoezy. We wczesnych postaciach (RA, RARS, RCMD, RCMD-RS; patrz klasyfikacja) bardziej nasilona jest zwykle apoptoza. W późnych stadiach (RAEB-1, RAEB-2) dominuje zwiększona proliferacja z wydłużonym czasem przeżycia. W tych postaciach częściej dochodzi do transformacji w ostre białaczki.
Wśród mechanizmów patogenetycznych prowadzących do rozwoju nieefektywnej hematopoezy postuluje się udział limfocytów T oraz wpływ nasilonej angiogenezy. Za przejście wczesnych form MDS w formy bardziej zaawansowane odpowiada skrócenie telomerów, nasilona metylacja oraz inaktywacja genu p15INK4b.
Objawy
Objawy nie są patognomoniczne dla zespołu mielodysplastycznego, mogą również występować w szeregu innych schorzeń.
W MDS dochodzi do rozwoju niedokrwistości, której objawami mogą być:
- osłabienie
- męczliwość
- bóle i zawroty głowy
- szybka akcja serca
- zmniejszenie koncentracji
- bladość skóry i śluzówek.
W przebiegu mielodysplazji może występować neutropenia. Zmniejszenie ilość granulocytów skutkuje ciężkimi zakażeniami bakteryjnymi i grzybiczymi.
Skutkiem małopłytkowości mogą być objawy skazy krwotocznej – wybroczyny na skórze oraz śluzówkach, a także groźne dla życia krwawienia, np. do centralnego systemu nerwowego.
Niekiedy występuje hepatomegalia, splenomegalia lub limfadenopatia (powiększenie węzłów chłonnych).
Diagnostyka
W diagnostyce zespołów mielodysplastycznych należy ocenić przede wszystkim krew obwodową, a także szpik (zarówno pod kątem morfologicznym, jak i cytogenetycznym).
W krwi obwodowej mogą występować następujące zaburzenia:
- niedokrwistość (najczęściej makrocytowa, rzadziej normocytowa)
- leukopenia (zmniejszenie całkowitej liczby białych ciałek krwi) z neutropenią (zmniejszenie liczby granulocytów)
- obecność blastów (niedojrzałych komórek układu hemopoetycznego) od 0% do 20% (wyższy procentowy odsetek świadczy już o białaczce)
- anomalia Pelgera-Hueta charakteryzująca się zmniejszeniem liczby segmentów jądra w komórkach neutrofilowych
- małopłytkowość
- retikulocytopenia (zmniejszenie liczby retikulocytów)
- niekiedy całkowita pancytopenia (zmniejszenie liczby wszystkich składników komórkowych krwi)
- nadmierna cytoliza erytrocytów w reakcji na składniki dopełniacza, spowodowane wadą w obrębie błony krwinki czerwonej.
Szpik w biopsji aspiracyjnej jest zazwyczaj bogatokomórkowy lub normokomórkowy z zaburzeniami hemopoezy różnych linii komórkowych: nieprawidłowe dojrzewanie jądra komórkowego i cytoplazmy oraz nieprawidłowe rozmieszczenie ziarnistości, obecność paraerytroblastów, mikromegakariocytów, megakariocytów jednojądrowych, zwiększona procentowa zawartość blastów.
W trepanobiopsji, oprócz zaburzonej hematopoezy, nieprawidłowe rozmieszczenie komórek prekursorowych oraz niekiedy cechy włóknienia szpiku.
Badanie cytochemiczne obrazuje obecność syderoblastów pierścieniowatych oraz tzw. syderoblastów patologicznych, tj. zawierających powyżej 5 ziarnistości żelazowych w cytoplazmie.
Na podstawie badania cytogenetycznego można stwierdzić nieprawidłowości w postaci: del(5q), monosomii 7, del(7q).
U osób z zespołem mielodysplastycznym może również występować zwiększone stężenie żelaza oraz hemoglobiny płodowej w surowicy.
Klasyfikacja
W użyciu są dwie klasyfikacje. Starsza – klasyfikacja FAB, powstała na zasadzie konsensusu, jaki wypracowała grupa amerykańskich, brytyjskich i francuskich badaczy. Klasyfikacja ta została po raz pierwszy opublikowana w 1976 roku, a następnie uaktualniona w 1982 roku. MDS został w niej podzielony na 5 grup. W 1985 roku na skutek obaw przed niedodiagnozowaniem ostrej białaczki szpikowej podtypu M6 (ostrej erytroleukemii; AML M6) klasyfikacja uległa dalszej modyfikacji.
Młodsza klasyfikacja została wprowadzona pod koniec lat 90. XX wieku przez grupę patologów i klinicystów pracujących pod auspicjami Światowej Organizacji Zdrowia (WHO). Stanowi zmodyfikowaną klasyfikację FAB.
Klasyfikacja FAB
Klasyfikacja FAB opiera się procentowej zawartości blastów w krwi obwodowej i szpiku kostnym oraz charakterystyce morfologicznej.
- RA (Refractory Anemia; niedokrwistość oporna na leczenie) – obecność < 1% blastów w krwi obwodowej i < 5% blastów w szpiku kostnym. ICD-O9980/3
- RARS (Refractory Anemia with Ringed Sideroblasts; niedokrwistość oporna na leczenie z obecnością pierścieniowatych syderoblastów) – obecność < 1% blastów w krwi obwodowej i < 5% blastów w szpiku kostnym, przy czym wśród blastów szpikowych > 15% stanowią tzw. syderoblasty pierścieniowate – erytroblasty, w których ziarna żelaza otaczają ponad 1/3 obwodu jądra. ICD-O9982/3
- RAEB (Refractory Anemia with Excess Blasts; niedokrwistość oporna na leczenie z nadmiarem blastów) – obecność < 5% blastów w krwi obwodowej i 5 – 20% blastów w szpiku kostnym. ICD-O9983/3
- RAEB-T (Refractory Anemia with Excess Blasts in Transformation; niedokrwistość oporna na leczenie z nadmiarem blastów w okresie transformacji) – obecność > 5% blastów w krwi obwodowej i 21 – 29% blastów w szpiku kostnym. Obecnie klasyfikuje się ją jako ostrą białaczkę szpikową. ICD-O9984/3
- CMML (Chronic myelomonocytic leukemia; przewlekła białaczka mielomonocytowa) – obecność < 5% blastów w krwi obwodowej i < 20% blastów w szpiku kostnym oraz > 1000/μl monocytów w krwi obwodowej. Obecnie jest klasyfikowana jako zespół mielodysplastyczno-mieloproliferacyjny. ICD-O9945/3
| Typ MDS | Blasty w krwi obwodowej % |
Blasty w szpiku kostnym % |
Mediana przeżycia (miesiące) |
Uwagi |
|---|---|---|---|---|
| RA | < 1 | < 5 | 30 – 65 | |
| RARS | < 1 | < 5 | 34 – 83 | Obecność > 15% syderoblastów pierścieniowatych w szpiku. |
| RAEB | < 5 | 5 – 20 | 8 – 18 | |
| RAEB-T | ≥ 5 | 21 – 29 | 4 – 11 | |
| CMML | < 5 | ≤ 20 | 15 – 32 | Obecne monocyty w krwi obwodowej w liczbie > 1000/μl. |
W 1985 roku wprowadzono modyfikację klasyfikacji w celu odróżnienia pacjentów z MDS a ostrą białaczką szpikową. U chorych z obecnością ≤ 50% erytroblastów w szpiku bierze się pod uwagę procentową zawartość wszystkich jądrzastych komórek blastycznych. Obecność ≥ 30% takich blastów upoważnia do rozpoznania ostrej erytroleukemii (AML M6). < 30% jest traktowane jako MDS.
Z chwilą występowania > 50% erytroblastów w szpiku, drugim krokiem diagnostycznym jest określenie występowania procentowej zawartości blastów wszystkich komórek jądrzastych z wyjątkiem tych z linii erytroidalnej. Przy obecności ≥ 30% takich komórek w różnicowaniu należy wziąć pod uwagę podtypy M1-M5 ostrej białaczki szpikowej. < 30% przemawia za rozpoznaniem zespołu mielodysplastycznego.
Klasyfikacja FAB była używana przez 20 lat. Obecnie, po wprowadzeniu przez WHO nowszej klasyfikacji, straciła na znaczeniu.
Klasyfikacja WHO
W 1999 roku wprowadzono modyfikację klasyfikacji FAB. Wprowadzono, między innymi, nową grupę schorzeń – RCMD (Refractory Cytopenia with Multilineage Dysplasia), która obejmowała pacjentów z patologicznymi zmianami nieograniczonymi tylko do linii erytroidalnej oraz zespół 5q-. RAEB-T klasyfikowana poprzednio jako zespół mielodysplastyczny została przesunięta do białaczek. Cezura w postaci procentowej zawartości blastów w szpiku została przesunięta z poprzednich 30% do 20%. Od tej granicy rozpoznaje się już białaczkę.
| Typ | Mediana przeżycia (miesiące) |
Transformacja w AML (% w okresie 5 lat) |
|---|---|---|
| RA | 67 | 7 |
| RARS | 70 | 1 |
| RCMD | 33 | 19 |
| RCMD-RS | 30 | 18 |
| RAEB-1 | 15 | 35 |
| RAEB-2 | 10 | 51 |
| MDS-U | ?? | ?? |
| MDS (del 5q) | 116 | 8 |
Klasyfikacja obejmuje następujące typy:
-
RA – Refractory Anemia:
- krew obwodowa: niedokrwistość; blasty < 1%; monocyty < 1000/μl
- szpik kostny: dysplazja tylko linii erytroidalnej; blasty < 5%; bez syderoblastów pierścieniowatych
-
RARS – Refractory Anemia with Ringed Sideroblasts
- krew obwodowa: niedokrwistość; blasty < 1%; monocyty < 1000/μl
- szpik kostny: dysplazja tylko linii erytroidalnej; blasty < 5%; ≥ 15% syderoblastów pierścieniowatych
-
RCMD – Refractory Cytopenia with Multilineage Dysplasia
- krew obwodowa: bicytopenia (zmniejszenie ilości 2 linii komórkowych) albo pancytopenia (niedobór wszystkich 3 linii komórkowych); blasty < 1%; monocyty < 1000/μl
- szpik kostny: dysplazja ≥ 10% komórek w 2≥ liniach; blasty < 5%; bez pałeczek Auera; < 15% syderoblastów pierścieniowatych
-
RCMD-RS – Refractory Cytopenia with Multilineage Dysplasia and Ringed Sideroblasts
- analogicznie jak w RCMD, tylko w szpiku obecnych ≥ 15% syderoblastów pierścieniowatych
-
RAEB-1 – Refractory Anemia with Excess Blasts 1
- krew obwodowa: cytopenie; blasty < 5%; monocyty < 1000/μl
- szpik kostny: dysplazja ≥ 1 linii; blasty 5 – 9%; bez pałeczek Auera
-
RAEB-2 – Refractory Anemia with Excess Blasts 2
- krew obwodowa: cytopenie; blasty 5 – 19%; obecne lub nie pałeczki Auera
- szpik kostny: dysplazja ≥ 1 linii; blasty 10 – 19%; możliwa obecność pałeczek Auera
-
MDS-U – Myelodysplastic Syndrome Unclassified
- krew obwodowa: cytopenie; blasty < 1%; obecne lub nie pałeczki Auera
- szpik kostny: dysplazja jednej linii; blasty < 5%; bez pałeczek Auera
-
MDS z izolowaną delecją 5q-
- krew obwodowa: niedokrwistość; prawidłowa liczba płytek lub trombocytoza
- szpik kostny: megakariocyty z jądrami o zmniejszonej liczbie płatów – prawidłowa lub zwiększona liczba; blasty < 5%; bez pałeczek Auera; izolowana delecja 5q.
Leczenie
Model terapii zależny jest od wieku i stanu ogólnego pacjenta (ocenianego na podstawie skali ECOG), oraz od klasyfikacji rokowniczej IPSS (patrz niżej):
- stan ciężki – leczenie łagodne
- stan dobry:
- chorzy w podeszłym wieku (> 60 r.ż.) – leczenie łagodne, w razie braku kwalifikacji do przeszczepu – leki demetylujące
- chorzy młodsi (< 60 r.ż.)
- ryzyko małe lub pośrednie – 1 według IPSS – leczenie łagodne
- ryzyko pośrednie – 2 lub duże – leczenie intensywne
leczenie łagodne – leczenie prowadzone w warunkach ambulatoryjnych (czynnik wzrostu, chemioterapia w małych dawkach, leczenie wspomagające)
leczenie intensywne – leczenie prowadzone w warunkach szpitalnych (chemioterapia skojarzona, przeszczepienie szpiku kostnego), w razie braku kwalifikacji do przeszczepu – leki demetylujące
Przeszczepienie szpiku
Jest to metoda z wyboru u chorych poniżej 45 roku życia (przy obecności zgodnego dawcy). Jest metodą umożliwiającą wyleczenie.
Chemioterapia
W leczeniu łagodnym zespołów mielodysplastycznych stosuje się małe dawki arabinozydu cytozyny (Ara-C).
W chemioterapii intensywnej można wykorzystywać modele stosowane w leczeniu ostrej białaczki szpikowej, np. model 3+7 polegający na podawaniu przez 7 dni Ara-C oraz przez 3 dni antracykliny (np. daunorubicyny). Do całkowitej remisji dochodzi u około 50% pacjentów, przy czym czas trwania remisji wynosi około 1 rok. Śmiertelność w trakcie chemioterapii szacuje się na 15%.
Leczenie wspomagające
W leczeniu wspomagającym stosuje się głównie uzupełnianie niedoborów elementów morfotycznych krwi:
- przetaczanie krwi – zazwyczaj stosuje się preparaty ubogoleukocytarne (zmniejszenie ryzyka powikłań)
- podawanie erytropoetyny (niekiedy w skojarzeniu z G-CSF) – najlepszą odpowiedź uzyskuje się w przypadku RARS
- podawanie G-CSF – leczenie neutropenii, przeciwdziałanie zakażeniom
- przetaczanie krwinek płytkowych w przypadku obecności skazy krwotocznej.
Leki immunomodulujące
W leczeniu MDS z izolowaną delecją 5q stosuje się lenalidomid.
Leki demetylujące
- 5-azacytydyna i decytabina – inhibitory metylacji DNA. Wykazują aktywność w MDS zwiększonego ryzyka. W badaniach z użyciem tych inhibitorów stwierdzono całkowitą remisję u 10% – 37% pacjentów oraz częściową remisję u 17% – 25% pacjentów. 5-azacytydydyna dwukrotnie zwiększa 2-letnie przeżycie oraz zwiększa o 50% medianę czasu przeżycia .
Leki eksperymentalne
ATG, trójtlenek arsenu, bewacizumab, deferasirox, varinostat, klaforabina [ASH, 2008]
Rokowanie
W 1997 roku wprowadzono do użytku punktowy system oceny pacjentów z zespołem mielodysplastycznym stratyfikujący ryzyko transformacji w ostrą białaczkę i oceniający medianę przeżycia (International Prognostic Scoring System). Wzięto w nim pod uwagę procentową zawartość blastów w szpiku, kariotyp oraz liczbę cytopenii (zmniejszenie ilości morfotycznych składników krążących w krwi obwodowej). Powyższe czynniki przedstawiono w systemie punktowym (od 0 do 2,0; licząc co pół punktu), przyporządkowując poszczególnej punktacji stosowne ryzyko:
| Punktacja |
Mediana przeżycia (lata) |
Transformacja w AML (%) |
|---|---|---|
| 0 | 5,7 | 19 |
| 0,5 – 1,0 | 3,5 | 30 |
| 1,5 – 2,0 | 1,2 | 65 |
| ≥ 2,5 | 0,4 | 100 |
| Małe ryzyko | ||
| Pośrednie ryzyko (1) | ||
| Pośrednie ryzyko (2) | ||
| Duże ryzyko | ||
- procentowa zawartość blastów w szpiku kostnym:
- 0,0 pkt – <5%
- 0,5 pkt – 5 – 10%
- 1,5 pkt – 11 – 20%
- 2,0 pkt – 21 – 30%
- kariotyp:
-
0,0 pkt – kariotyp korzystny:
- normalny
- -Y
- del(5q)
-
0,5 pkt – kariotyp pośredni:
- wszelkie pozostałe anomalie niewymienione przy kariotypie niekorzystnym (patrz niżej)
-
1,0 pkt – kariotyp niekorzystny:
- złożone defekty (≥3)
- anomalie chromosomu 7
-
0,0 pkt – kariotyp korzystny:
- cytopenia (neutrofile < 1800/μl; Hb < 10 g/dl; PLT < 100 000/μl):
- 0,0 pkt – 0 – 1
- 0,5 pkt – 2 – 3
W celu określenia kategorii ryzyka sumuje się punkty (patrz tabelka obok). Kategorie IPSS wykorzystuje się również w celu wyboru stosownego algorytmu leczenia (patrz sekcja leczenie).
Historia
Po raz pierwszy MDS został opisany w 1907 roku przez Luzatto. Był to przypadek przewlekłej anemii związanej z hiperplazją szpikowych komórek erytroidalnych. Luzatto określił to schorzenia jako anemię pseudoaplastyczną. Następnie dzięki badaniom Rhoadsa i Bomforda do użytku weszło określenie refractory anemia (anemia oporna). W miarę zwiększania ilości doniesień o możliwości tranformacji zespołów mielodysplastycznych w ostre białaczki, na początku lat 50. XX wieku Block i Hamilton zaproponowali określenie preleukemia. W 1956 roku wydzielono z grupy pacjentów z anemią oporną osoby z obecnością w szpiku syderoblastów pierścieniowatych. Na podstawie tych doniesień William Dameshek wysunął podejrzenie, że anemia syderoblastyczna może stanowić wczesną formę erytroleukemii.
Określenie preleukemia (stan przedbiałaczkowy) szybko okazało się błędne, gdyż wielu pacjentów u których rozpoznano ten stan nie rozwinęło białaczki. Ostateczne uporządkowanie kwestii klasyfikacji i nazewnictwa zespołów mielodysplastycznych nastąpiło w 1976 roku, kiedy to międzynarodowy zespół badaczy (FAB) ustalił konsensus w tej sprawie. Wprowadzono wówczas klasyfikacja funkcjonowała przez ponad 20 lat, aż do czasu kiedy Światowa Organizacja Zdrowia w 1999 roku wprowadziła uaktualnienia, które funkcjonują do dzisiaj.
Historyczne nazewnictwo zespołów mielodysplastycznych: stan przedbiałaczkowy, niedokrwistość oporna na leczenie, niedokrwistość oporna na leczenie z nadmiarem komórek blastycznych, tląca się ostra białaczka, przewlekła mieloza erytemiczna, podostra białaczka mielomonocytowa, podostra białaczka szpikowa, hipoplastyczna ostra białaczka szpikowa, dysplazja hematopoetyczna.
Bibliografia
- Andrzej Szczeklik: Choroby wewnętrzne. Kraków: Medycyna Praktyczna, 2005, s. 1483-1488. ISBN 83-7430-031-0.
- Lewis R. Silverman: Myelodysplastic Syndrome. [dostęp 2012-09-14]. (ang.).