
| acrocephalosyndactylia | |
| Klasyfikacje | |
| ICD-10 | |
|---|---|
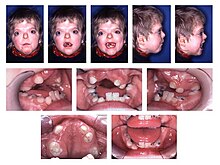
Zespół Aperta (łac. acrocephalosyndactylia, ang. Apert syndrome) – uwarunkowany genetycznie zespół wad wrodzonych, obejmujący wrodzone zarośnięcie szwów czaszkowych oraz palcozrost. Większość przypadków jest sporadyczna, ale stwierdzono rodzinne występowanie choroby dziedziczonej autosomalnie dominująco. Zespół Aperta wywołują mutacje w genie receptora 2 czynnika wzrostu fibroblastów FGFR2 (Fibroblast Growth Factor Receptor 2) w locus 10q26. Zespół Crouzona i zespół Pfeiffera o fenotypach częściowo pokrywających się nawzajem i z fenotypem zespołu Aperta są spowodowane mutacjami w tym samym genie, są to więc schorzenia alleliczne.
Objawy i przebieg
Na obraz choroby składają się deformacje czaszki związane z przedwczesnym zarośnięciem szwów czaszkowych (craniosynostosis). Towarzyszą im zrośnięcie palców 2–5 (syndaktylia) rąk i stóp. Zmiany w obrębie twarzoczaszki prowadzą do pseudoprognatyzmu wskutek względnej hipoplazji szczęki i innych struktur kostnych środkowej części twarzy. Wstępuje hiperteloryzm oczny. Rozszczep podniebienia spotykany jest u około 25% chorych. U około 38% pacjentów występuje niepełnosprawność intelektualna (31% w stopniu lekkim, 7% w stopniu głębokim).
Historia
Chorobę opisał jako pierwszy francuski pediatra Eugène Apert (1868-1940) w 1906 roku. Genetyczne podłoże choroby wyjaśniła praca Andrew O. M. Wilkiego i wsp. z 1995 roku.
Linki zewnętrzne
- APERT SYNDROME w bazie Online Mendelian Inheritance in Man (ang.)
- Apert’s syndrome w bazie Who Named It (ang.)