
| syndroma Ehlers-Danlos | |
| |
| Klasyfikacje | |
| ICD-10 | |
|---|---|

Zespół Ehlersa-Danlosa, EDS (od ang. Ehlers-Danlos syndrome) – grupa chorób genetycznych charakteryzująca się nadmierną wiotkością (hipermobilnością) stawów, delikatną oraz (jedynie w niektórych typach) hiperelastyczną skórą. Zaburzenie dotyczy nieprawidłowości w syntezie i/lub budowie tkanki łącznej, czyli głównej tkanki budulcowej organizmu, co oznacza, że objawy mogą obejmować różne organy i układy ciała chorego. Aktualnie wyróżnia się 14 typów EDS, z czego tylko typ hipermobilny (5) nie ma jeszcze określonej mutacji genetycznej i diagnozę hEDS stawia się jedynie na podstawie objawów klinicznych oraz wykluczenia innych zaburzeń tkanki łącznej. Symptomy u różnych chorych wahają się od lekkich do bardzo ciężkich. Notowane są też przypadki śmiertelne (szczególnie osób z vEDS i kEDS, ale mogą dotyczyć każdego typu).
Historia
Pierwsze doniesienia o wiotkości stawowej pojawiają się w pracy Hipokratesa "O powietrzu, wodach i okolicach" z ok. 400 r. p.n.e. odnośnie do ludu Scytów.
W 1682 roku, holenderski chirurg Job Janszoon van Meekeren napisał: w 1657 widzieliśmy w naszym szpitalu [...] młodego Hiszpana o imieniu Georg Albes, który swoją lewą ręką schwycił skórę ponad kością ramienną i naciągnął ją prawie do ust. Potem skóra powoli wróciła do swojego naturalnego położenia.
W 1892 zespół opisał Mikołaj Aleksandrowicz Czernogubow, a na początku XX w., niezależnie od siebie, Edvard Lauritz Ehlers i Henri-Alexandre Danlos. Rodzinne występowanie choroby odnotował Johnson w 1949 r. Jako pierwszy odmienne typy choroby wyodrębnił Barabas w 1967.
W 1988 r. Zespół Ehlersa Danlosa podzielono na 11 typów – klasyfikacja berlińska.
W 1998 r. powstała klasyfikacja Villefranche wyróżniająca 6 typów.
W 2017 r. w związku z rozwojem badań genetycznych (NGS) sklasyfikowano 13 typów Zespółu Ehlersa Danlosa, wraz z wyszczególnieniem mutacji genetycznych. Jedynie typ hipermobilny (aktualnie 5, wcześniej typ III) ma wciąż nieokreśloną mutację genetyczną, a diagnozę stawia się na podstawie objawów klinicznych oraz wykluczenia innych chorób tkanki łącznej. Kilka miesięcy później dodano nowy (14) typ EDS, dotyczący jedynie 4 osób z 3 niespokrewnionych ze sobą rodzin.
Etiologia
Podłożem zaburzeń obserwowanych w EDS są nieprawidłowości w syntezie lub obróbce potranslacyjnej kolagenu, jak również macierzy międzykomórkowej tkanki łącznej, a także dysfunkcje biosyntezy glikozoaminoglikanów, układu dopełniacza i procesów wewnątrzkomórkowych. Defekty są wynikiem mutacji genetycznych; dziedziczenie, w zależności od typu, jest autosomalne dominujące lub recesywne.
Czasem też pojawia się mutacja de novo. Oznacza to, że błąd w genie występuje tylko u dziecka, natomiast rodzice są zdrowi i nie są nosicielami wadliwego genu.
Częstość występowania
Szacuje się, że na Zespół Ehlersa-Danlosa choruje 1:2500 – 1:10 000 osób. Rozbieżność wynika z braku świadomości choroby wśród lekarzy oraz rzadkiego jej diagnozowania.
Obraz kliniczny


Aktualnie opisano 14 typów Zespołu Ehlersa Danlosa.
Objawy podstawowe
- stawowe: hipermobilność (według skali Beightona), luźność/niestabilność/wiotkość stawów, prowadząca do ich dyslokacji i/lub podwichnięć; zakres ruchu stawów jest wyraźnie większy niż normalnie (bez konieczności ćwiczeń rozciągających), wcześnie pojawiają się też zwyrodnienia stawów oraz powikłania po licznych urazach;
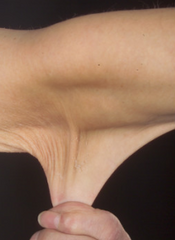
- skórne: skóra jest delikatna, jedwabista, w niektórych typach również rozciągliwa; po urazach gojenie trwa dłużej i nierzadko powstają bliznowce; występuje też skłonność do łatwego siniaczenia nawet przy niewielkim urazie.
Objawy dodatkowe
- niedomykalność zastawki mitralnej serca (czasem też innych);
- delikatność i kruchość narządów (np. jelit – IBS, pęcherza – nadmierna rozciągliwość ścian pęcherza i/lub neurogenny pęcherz, naczyń krwionośnych – niewydolność naczyń krwionośnych, POTS, żylaki);
- może dojść do pęknięcia dużych naczyń krwionośnych oraz narządów (głównie w typie vEDS);
- problemy ze wzrokiem (np. astygmatyzm, trudność w dobraniu odpowiednich okularów, z uwagi na zmienność wady wzroku);
- szpotawość lub koślawość stóp i/lub kolan (nierzadko od urodzenia);
- skolioza i/lub kifoskolioza (duże skrzywienia występują głównie w typie kEDS);
- inne.
Skala Beightona
W drugiej połowie XX w. prof. Peter Beighton opracował listę 5 czynności, które miały na celu potwierdzenie lub wykluczenie zespołu hipermobilności. Są to:
- przeprost w stawie kolanowym powyżej 10°
- przeprost w stawie łokciowym powyżej 10°
- możliwość biernego przyciągnięcia kciuka do przedramienia
- możliwość biernego wygięcia palców dłoni w stronę grzbietową, równolegle do przedramienia
- możliwość położenia płasko dłoni na podłodze przy głębokim skłonie przy wyprostowanych stawach kolanowych.
Każdy staw liczony jest oddzielnie, co oznacza, że maksymalnie można otrzymać 9 punktów. Do postawienia diagnozy o zespole nadmiernej ruchomości stawów (ang. BJHS – benign joint hypermobility syndrome lub JHS – joint hypermobility syndrome), pacjent musi uzyskać przynajmniej 5 punktów.
Typy Zespołu Ehlersa – Danlosa
| Nr. | Typ kliniczny | Skrót | Dziedziczenie | Gen | Locus | Białko | OMIM |
|---|---|---|---|---|---|---|---|
| 1. | Klasyczny | cEDS | AD | COL5A1
COL5A2 COL1A1 |
3q34.3
2q32.2 17q21.33 |
Kolagen typu V
Kolagen typu V Kolagen typu I (p. Arg312Cys) |
130000 |
| 2. | Jak klasyczny | clEDS | AR | TNXB | 6p21.33-p21.32 | Tenascyna XB | 600985 |
| 3. | Sercowo – zastawkowy | cvEDS | AR | COL1A2 | 7q21.3 | Kolagen typu I
(Bialleliczna mutacja, która prowadzi do COL1A2 NMD i całkowitego braku łańcucha α2(1) kolagenu) |
225320 |
| 4. | Naczyniowy | vEDS | AD | COL3A1
COL1A1 |
2q32.2
17q21.33 |
Kolagen typu III
Kolagen typu I |
130050 |
| 5. | Hipermobilny | hEDS | AD | ? | ? | ? | 130020 |
| 6. | Artrochalasia | aEDS | AD | COL1A1
COL1A2 |
17q21.33
7q21.3 |
Kolagen typu I
– |
130060 |
| 7. | Skórny | dEDS | AR | ADAMTS2 | 5q35.3 | ADAMTS-2 | 225410 |
| 8. | Kifoskoliotyczny | kEDS | AR | PLOD1
FKBP14 |
1p36.2
7p14.3 |
HYDROKSYLAZA LIZYLOWA I
FKBP22 |
225400 |
| 9. | Zespół Kruchej Rogówki | BCS | AR | ZNF469
PRDM5 |
16q24
4q27 |
ZNF469
PRDM5 |
229200 |
| 10. | Spondylodysplastyczny | spEDS | AR | B4GALT7
B3GALT6 |
5q35.3
1p36.33 |
galaktozylotransferaza
beta4GalT7 galaktozylotransferaza II beta3GalT6 |
130070 |
| 11. | Z przykurczami mięśniowymi | mcEDS | AR | CHST14
DSE |
15q15.1
6q22.1 |
Dermatan-4-sulfotransferaza-1
Dermatan sulfate epimeraza-1 |
601776 |
| 12. | Miopatyczny | mEDS | AD lub AR | COL12A1 | 6q13-q14 | XII | 616471 |
| 13. | Okołozębowy | pEDS | AR | C1R
C1S |
12p13.31 |
C1R
C1S |
130080 |
| 14. | (Brak nazwy) | --- | AR | AEBP1 | ACLP |
Typ klasyczny (cEDS, ang. classical EDS)
Kryteria większe:
I Kryterium: rozciągliwość skóry (1, 2) i blizny atroficzne,
II Kryterium: ogólna wiotkość stawowa (GJH).
Kryteria mniejsze:
- łatwe siniaczenie,
- aksamitna skóra,
- kruchość i pękanie skóry,
- pojawianie się pseudoguzów (łokcie, palce, okolice blizn),
- występowanie sferoidów (małych, twardych, okrągłych, przesuwalnych tworów; głównie na przedramionach),
- przepukliny,
- fałdy nakątne (wewnętrzne kąciki oczu),
- powikłania wiotkości stawów (np. skręcenia, zwichnięcia, podwichnięcia, ból, płaskostopie elastyczne),
- historia rodzinna: krewni I stopnia spełniający kryteria EDS.
Minimalne kryteria sugerujące cEDS obejmują:
- I kryterium większe (rozciągliwość skóry)
- II kryterium większe i/lub minimum trzy kryteria mniejsze.
Klasyczny typ EDS jest związany z mutacjami genów: COL5A1, COL5A2 lub – rzadziej – COL1A2, powodującymi uszkodzenia kolagenu typu V lub I.
Sposób dziedziczenia: autosomalny dominujący.
Typ podobny do klasycznego (clEDS, ang. classical-like EDS)
Kryteria większe:
I Kryterium: rozciągliwa, aksamitna skóra, bez atroficznych blizn,
II Kryterium: wiotkość stawów ze zwichnięciami lub bez nich,
III Kryterium: spontaniczne siniaczenie, pojawianie się wybroczyn.
Kryteria mniejsze:
- zniekształcenie stóp: poszerzenie/opuchnięcie przodostopia, brachydaktylia z nadmierną ilością skóry, płaskostopie, halluksy, grudki piezogeniczne,
- obrzęki nóg bez niewydolności serca,
- łagodne osłabienie mięśniowe,
- polineuropatia aksonalna,
- atrofia mięśni dłoni i stóp,
- akrogeria dłoni i stóp, palec/palce malleta, klinodaktylia, brachydaktylia,
- wypadanie pochwy, macicy, odbytu.
Minimalne kryteria sugerujące clEDS obejmują:
- I, II i III Kryterium większe
- pozytywny wynik badania genetycznego clEDS w wywiadzie rodzinnym.
Podobny do klasycznego typ EDS jest związany z mutacją genu TNXB, który powoduje uszkodzenie białka, zwanego tenascyną XB.
Sposób dziedziczenia: autosomalny recesywny.
Typ sercowo-zastawkowy (cvEDS, ang. cardiac-valvular EDS)
Kryteria większe:
I Kryterium: ciężkie, postępujące problemy sercowo – zastawkowe (niewydolność zastawki mitralnej, niewydolność zastawki aortalnej),
II Kryterium: skóra cienka, rozciągliwa, z tendencją do siniaczeń i uszkodzeń; blizny atroficzne,
III Kryterium: wiotkość stawowa (uogólniona lub dotycząca małych stawów).
Kryteria mniejsze:
- przepuklina pachwinowa,
- zniekształcenie klatki piersiowej (głównie wklęśnięcie),
- dyslokacje stawów,
- zniekształcenia stóp: płaskostopie, stopy płasko – koślawe, halluksy.
Minimalne kryteria sugerujące cvEDS obejmują:
- I kryterium większe (problemy sercowo-zastawkowe),
- pozytywny wynik badania genetycznego cvEDS w wywiadzie rodzinnym
- Jedno inne kryterium większe i/lub co najmniej 2 kryteria mniejsze.
CvEDS jest związany z brakiem pro-α2-chain w I typie kolagenu z powodu mutacji genu COL1A2, która prowadzi do zniszczenia mRNA, zawierającego przedwczesny kodon STOP.
Sposób dziedziczenia: autosomalny recesywny.
Typ naczyniowy (vEDS, ang. vascular EDS)

Kryteria większe:
I Kryterium: historia rodzinna obejmująca przypadki chorych na vEDS z udokumentowaną mutacją genu COL3A1,
II Kryterium: pęknięcie aorty w młodym wieku (<40 r.ż.),
III Kryterium: samoistne pęknięcie esicy bez stwierdzonych uchyłków lub innych patologii jelit,
IV Kryterium: pęknięcie macicy w trzecim trymestrze ciąży – w historii pacjentki brak cesarskiego cięcia i/lub poważnego pęknięcia krocza,
V Kryterium: przetoka szyjno – jamista (carotid-cavernous sinus fistula) – pomiędzy tętnicą szyjną a zatoką jamistą – bez urazu.
Kryteria mniejsze:
- pojawianie się siniaków bez urazu lub w nietypowych miejscach jak policzki czy plecy,
- cienka, przezroczysta skóra z widocznymi naczyniami,
- charakterystyczny wygląd twarzy (cienki, “wychudły” nos, cienkie wargi, zapadnięte policzki, wytrzeszcz oczu),
- spontaniczna odma opłucnowa,
- akrogeria (starczy wygląd dłoni, stóp),
- stopy końsko-szpotawe,
- wrodzona dyslokacja stawów biodrowych,
- wiotkość małych stawów,
- pękanie ścięgien i mięśni,
- keratoconus (stożek rogówki),
- nieprawidłowości dziąseł (kruchość, zanik),
- wczesne żylaki (przed 30 rokiem życia i u kobiet, które nie rodziły).
Minimalne kryteria sugerujące vEDS obejmują:
- występowanie choroby w rodzinie,
- pęknięcia lub rozwarstwienia tętnic u osób poniżej 40 roku życia,
- niewyjaśnione pęknięcia esicy.
LUB
- Spontaniczna odma płucna występująca z innymi objawami vEDS
LUB
- Badanie genetyczne pod kątem vEDS powinny być rozważone również u pacjenta spełniającego kombinację kryteriów mniejszych.
Pacjenci z vEDS najczęściej wykazują mutację w genie COL3A1, z rzadkim wyjątkiem specyficznej mutacji substytucji argininy na cysteinę w genie COL1A1, która również powoduje kruchość naczyń i może naśladować COL3A1-vEDS. Bardzo rzadko można zidentyfikować bialleliczny wariant w genie COL3A1.
Sposób dziedziczenia: autosomalny dominujący.
Typ hipermobilny (hEDS, ang hypermobile EDS)
W przypadku hEDS, diagnoza jest stawiana wyłącznie na podstawie objawów klinicznych. Nie ma badań genetycznych, które mogłyby jednoznacznie potwierdzić rozpoznanie. Dlatego istotnym jest, aby umieć odróżnić hEDS od innych zaburzeń tkanki łącznej, zwłaszcza od izolowanych typów wiotkości stawowej.
Kryteria diagnostyczne:
I Kryterium: uogólniona wiotkość stawowa (GJH),
II Kryterium: co najmniej 2 z 3 poniższych cech (A,B,C):
A. Zaburzenia w budowie tkanki łącznej dotyczące całego ciała (co najmniej 5 z poniższych objawów):
- wyjątkowo delikatna lub aksamitna skóra;
- łagodna rozciągliwość skóry;
- niewyjaśnione rozstępy (bezbarwne lub czerwone) na plecach, udach, piersiach i/lub brzuchu, występujące u dojrzewających dzieci, mężczyzn, dziewczynek przed okresem dojrzewania bez historii związanej z wyraźnym wzrostem lub spadkiem wagi;
- obustronne grudki piezogeniczne na spodniej części stóp – przepukliny tłuszczu podskórnego;
- liczne, nawracające przepukliny brzuszne;
- blizny atroficzne (przynajmniej w dwóch różnych miejscach, jednak bez cech tworzenia bliznowców jak w typie klasycznym);
- wypadanie narządów (odbytu, pochwy) u dzieci, mężczyzn lub kobiet, które nie rodziły, bez chorobliwej otyłości lub innych zaburzeń klinicznych, mogących do tego predysponować;
- stłoczenie zębów oraz wąskie i wysokie podniebienie (tzw. “Gotyckie podniebienie”);
- arachnodaktylia (długie, cienkie palce, pozytywny objaw Steinberga i/lub objaw Walkera po obu stronach);
- zasięg ramion większy niż wzrost >1,05;
- wypadanie płatka zastawki mitralnej – delikatne lub mocniejsze według kryteriów echokardiografii;
- poszerzenie opuszki aorty w skali Z-score >+2;
B. w wywiadzie rodzinnym występuje przynajmniej jedna osoba z pokrewieństwa pierwszego stopnia, spełniająca kryteria diagnostyczne dla hEDS,
C. problemy mięśniowo-szkieletowe (co najmniej 1 z wymienionych poniżej):
- ból mięśniowo-szkieletowy (przynajmniej w jednej kończynie, pojawiający się codziennie, przez okres minimum 3 miesięcy);
- chroniczny, rozległy ból, trudny do zlokalizowania, trwający przez okres minimum 3 miesięcy;
- nawracające, atraumatyczne dyslokacje stawów lub swobodna niestabilność stawów (a lub b):
a) więcej, niż trzy atraumatyczne dyslokacje tego samego stawu lub więcej niż dwie atraumatyczne dyslokacje w dwóch różnych stawach pojawiające się w różnych odstępach czasu;
b) medyczne potwierdzenie niestabilności w przynajmniej dwóch różnych stawach pojawiające się bez urazu.
III Kryterium: wszystkie wymienione poniżej warunki muszą być spełnione:
- brak szczególnie delikatnej skóry, która mogłoby skłaniać do postawienia diagnozy o innych typach EDS;
- wykluczenie innych – wrodzonych i nabytych – zaburzeń tkanki łącznej, w tym autoimmunologicznych zaburzeń reumatycznych; wykluczenie innych diagnoz, które obejmują hipermobilność stawów z powodu hipotonii mięśniowej i/lub luźności tkanki łącznej. Alternatywne diagnozy mogą, lecz nie muszą sugerować zaburzenia neuromięśniowe (np. EDS typ miopatyczny, miopatia Bethlema), inne zaburzenia hipermobilności stawów (np. inne typy EDS, zespół Loeysa-Dietza, zespół Marfana), i dysplazję szkieletową (np. OI). Wykluczenie tych zaburzeń opiera się na wywiadzie rodzinnym, badaniu lekarskim i/lub badaniu genetycznym. U pacjentów z nabytym zaburzeniem tkanki łącznej (np. toczeń, reumatoidalne zapalenie stawów), dodatkowa diagnoza o hEDS może być postawiona tylko wtedy, gdy pacjent spełnia cechy A i B z kryterium II. Cechy C kryterium II (chroniczny ból i/lub niestabilność) nie mogą być uznane za objawy hEDS, gdy pacjent posiada diagnozę innej choroby tkanki łącznej.
Inne objawy, które mogą pojawić się u osób z hEDS, to:
- zaburzenia snu,
- zmęczenie,
- posturalna tachykardia ortostatyczna,
- zaburzenia funkcji układu pokarmowego,
- dysautonomia,
- lęk
- depresja.
Jednak na dzień dzisiejszy, istnieje zbyt mało danych, które pozwalałyby uznać te dolegliwości za formalne kryteria diagnostyczne.
Typ artrochalasia (aEDS, ang. Arthrochalasia EDS)
Kryteria większe:
I Kryterium: wrodzone obustronne zwichnięcie bioder,
II Kryterium: ciężka wiotkość stawów z nawracającymi dyslokacjami/subluksacjami,
III Kryterium: rozciągliwość skóry.
Kryteria mniejsze:
- hipotonia mięśniowa,
- kifoskolioza,
- łagodna osteopenia w RTG,
- delikatność tkanek, atroficzne blizny,
- łatwe siniaczenie.
Minimalne kryteria sugerujące aEDS obejmują:
- I kryterium większe
- III kryterium większe I/LUB II kryterium większe i przynajmniej dwa kryteria mniejsze
Typ arthrochalasia EDS jest spowodowany mutacjami genów COL1A1 i COL1A2, uszkadzającymi kolagen typu I.
Sposób dziedziczenia: autosomalny dominujący.
Typ skórny (dEDS, ang. dermatosparaxis EDS)
Kryteria większe:
I Kryterium: ekstremalna delikatność skóry z wrodzonym lub poporodowym pękaniem,
II Kryterium: charakterystyczna dysmorfia twarzy (widoczna po urodzeniu lub pojawiająca się w dzieciństwie),
III Kryterium: hipertrofia skóry, która wręcz zwisa, z przerośniętymi fałdami skóry w okolicy nadgarstków i kostek,
IV Kryterium: nadmierne marszczenie się skóry na wewnętrznej części dłoni,
V Kryterium: poważna skłonność do siniaków, zwiększająca ryzyko podskórnych krwawień i naczyniaków,
VI Kryterium: przepuklina pępkowa,
VII Kryterium: niski wzrost,
VIII Kryterium: krótkie kończyny, dłonie, stopy,
IX Kryterium: komplikacje poporodowe związane z dużą delikatnością tkanki łącznej.
Kryteria mniejsze:
- delikatna skóra,
- rozciągliwa skóra,
- blizny atroficzne,
- ogólna wiotkość stawowa,
- zaburzenia związane z delikatnością narządów wewnętrznych (np. pęknięcie pęcherza moczowego, pęknięcie przełyku, wypadanie odbytu),
- opóźniony rozwój motoryczny,
- osteopenia,
- hirsutyzm (nadmierne owłosienie),
- nieprawidłowość w budowie zębów,
- zaburzenia refrakcji (krótkowzroczność, astygmatyzm),
- zez.
Minimalne kryteria sugerujące dEDS obejmują:
- I kryterium większe
- II kryterium większe
- Jedno kryterium większe i/lub 3 kryteria mniejsze
Typ skórny EDS powodowany jest bialleliczną mutacją na genie ADAMTS2. Jest to jedyny gen, odpowiedzialny za dEDS.
Sposób dziedziczenia: autosomalny recesywny.
Typ kifoskoliotyczny (kEDS, ang. kyphoscoliotic EDS)
Kryteria większe:
I Kryterium: wrodzona hipotonia mięśniowa;
II Kryterium: wrodzona lub wczesna kifoskolioza (postępująca lub nie);
III Kryterium: ogólna wiotkość stawowa (GJH) z dyslokacjami/subluksacjami (szczególnie ramion, bioder i kolan).
Kryteria mniejsze:
- rozciągliwa skóra,
- łatwość siniaczenia,
- pękanie lub tworzenie się tętniaków na średniej wielkości tętnicach,
- osteopenia/osteoporoza,
- niebieska białkówka oka,
- przepukliny (pępkowa lub pachwinowa),
- deformacja klatki piersiowej,
- cechy marfanoidalne,
- stopy końsko-szpotawe,
- zaburzenia refrackji (krótkowzroczność, dalekowzroczność),
Kryteria mniejsze specyficzne dla genu:
- PLOD1
- delikatna skóra (łatwo powstające siniaki, krucha skóra, trudno gojące się rany, atroficzne blizny, które się rozchodzą),
- delikatność/pękanie twardówki oka,
- rogówka mała (łac. microcornea),
- dysmorfia twarzy,
- FKBP14
- wrodzone upośledzenie słuchu (głuchota czuciowo-nerwowa, głuchota przewodzeniowa, mieszany typ głuchoty),
- rogowacenie mieszkowe (follicular hyperkeratosis),
- atrofia mięśni,
- uchyłek pęcherza.
Minimalne kryteria sugerujące kEDS obejmują:
- I kryterium większe – wrodzona hipotonia mięśniowa
- II kryterium większe – wrodzona lub wczesna kifoskolioza
- III kryterium większe – GHJ I/LUB 3 kryteria mniejsze (ogólne kryteria mniejsze lub gen-kryteria mniejsze)
U większość pacjentów z kEDS występuje mutacja genu PLOD1 lub FKBP14, które powodują zmiany w białkach LH1 i FKBP22.
Sposób dziedziczenia: autosomalny recesywny.
Zespół Kruchej Rogówki (BCS, ang. Brittle Cornea Syndrome)
Kryteria większe:
I Kryterium: cienka rogówka (grubość rogówki <400um), z lub bez wystąpienia pęknięcia rogówki,
II Kryterium: wcześnie pojawia się i postępuje stożek rogówki,
III Kryterium: wcześnie pojawiająca się i postępująca rogówka kulista,
IV Kryterium: niebieskie twardówki oka.
Kryteria mniejsze:
- wyłuszczenie oka lub pojawiające się bliznowacenia w oku związane z pęknięciami rogówki
- progresywne zmniejszanie grubości rogówki, głównie w centralnej części,
- duża krótkowzroczność, z normalnym lub lekko przesuniętym punktem ogniskowania,
- odwarstwienie rogówki,
- postępująca głuchota, często o typie mieszanym (czuciowo-nerwowym i przewodzeniowym), większa wrażliwość na wysokie dźwięki (sloping pure tone audiogram)
- nadmiernie elastyczna błona bębenkowa,
- dysplazja rozwojowa bioder,
- delikatna hipotonia u dzieci,
- skolioza,
- arachnodaktylia,
- hipermobilność stawów dalszych,
- płaskostopie, haluksy,
- delikatny przykurcz palców (głównie małych palców dłoni),
- delikatna, jedwabna, przezroczysta skóra.
Minimalne kryteria sugerujące BCS:
- I kryterium główne
- jeszcze jedno kryterium główne lub przynajmniej 3 kryteria mniejsze
BCS jest związany z mutacjami genów ZNF469 i PRDM5, powodującymi zmiany w białkach ZNF469 i PRDM5.
Mutacje innych genów u osób z klinicznymi objawami BCS: PLOD1; FKBP14; B4GALT7; B3GALT6; SLC39A13; CHST14 i DSE.
Brak mutacji w powyższych genach nie wyklucza diagnozy BCS, ponieważ badania standardowe mogą nie wykazać mutacji lub BCS może być związane z mutacjami innych, niezbadanych jeszcze genów.
Sposób dziedziczenia: autosomalny recesywny.
Typ spondylodysplastyczny (spEDS)
Kryteria większe:
I Kryterium: niski wzrost,
II Kryterium: hipotonia mięśniowa (waha się od ciężkiej hipotonii wrodzonej do delikatnej hipotonii pojawiającej się później),
III Kryterium: zniekształcenia, łukowatość kończyn.
Kryteria mniejsze:
- skóra hiperelastyczna, delikatna, przezroczysta, ciastowata,
- płaskostopie,
- opóźniony rozwój motoryczny,
- osteopenia,
- opóźniony rozwój poznawczy.
Kryteria mniejsze specyficzne dla genu:
B4GALT7
- kościozrost kości promieniowej i łokciowej,
- obustronny przykurcz w stawach łokciowych lub ograniczona ruchomość stawów łokciowych,
- ogólna wiotkość stawowa,
- pojedyncze odwrócenie bruzd na dłoniach,
- charakterystyczna budowa twarzy,
- charakterystyczne zmiany w obrazach radiologicznych,
- ciężka dalekowzroczność,
- zamglenie rogówki.
B3GALT6
- postępująca kifoskolioza (wrodzona lub pojawiająca się w dzieciństwie),
- ogólna lub zlokalizowana na stawach dalszych hipermobilność stawów, z dyslokacjami,
- przykurcze stawów (wrodzone i postępujące), głównie rąk,
- charakterystyczna budowa palców (palce są szczupłe, zwężające się, łopatkowate, szerokie na końcach, długie – arachnodaktylia),
- stopy końsko-szpotawe,
- charakterystyczna budowa twarzy,
- przebarwienie zębów, dysplastyczne zęby,
- charakterystyczny obraz radiograficzny,
- osteoporoza z wieloma samoistnymi złamaniami,
- tętniak aorty wstępującej,
- hipoplazja płuc, choroby związane z obniżoną pojemnością płuc.
SLC39A13
- wytrzeszcze oczu z niebieską rogówką,
- dłonie z drobnym pofałdowaniem części wewnętrznej,
- atrofia mięśni okolic kciuka wewnętrznej części dłoni, ze zwężającymi się palcami,
- hipermobilność stawów dalszych (dystalnych).
Minimalne kryteria sugerujące spEDS:
- I kryterium większe
- II kryterium większe
- Charakterystyczne zmiany w obrazie radiograficznym i przynajmniej 3 kryteria mniejsze (ogólne lub związane z genem)
Spondylodysplastyczny EDS jest związany z mutacjami genów B4GALT7, B3GALT6 i SLC39A13, powodującymi zmiany białek β4GalT7, β3GalT6 i ZIP13.
Mutacje innych genów u pacjentów wykazujących objawy kliniczne spEDS: PLOD1, FKBP14, ZNF469, PRDM5, CHST14 i DSE
Sposób dziedziczenia: autosomalny recesywny.
Typ z przykurczami mięśniowymi (mięśniowo-przykurczowy, mcEDS, ang. musculocontractural EDS)
Kryteria większe
I Kryterium: wrodzone przykurcze wielu mięśni przeciwstawnych, charakterystyczny przykurcz mięśni przywodziciela-zginacza i/lub stopa końsko-szpotawa,
II Kryterium: charakterystyczne zmiany twarzoczaszki, które widoczne są tuż po urodzeniu lub we wczesnym dzieciństwie,
III Kryterium: charakterystyczne zmiany skórne, jak np. nadmierna rozciągliwość skóry, łatwość siniaczenia, delikatność skóry z bliznami przerostowymi oraz zwiększona ilość fałdów na wewnętrznej stronie dłoni.
Kryteria mniejsze
- nawracające, chroniczne dyslokacje;
- deformacja klatki piersiowej (płaska, wklęsła);
- deformacje kręgosłupa (skolioza, kifoskolioza);
- szczególna budowa palców rąk (zwężające się ku końcowi, smukłe, cylindryczne);
- postępujące deformacje stóp (koślawe, płaskie, wysklepione);
- duże krwiaki śródskórne;
- chroniczne zaparcia;
- uchyłki jelita grubego;
- odma opłucnowa/odma opłucnowa z krwiakiem;
- kamica nerkowa/kamica żółciowa;
- wodonercze;
- wnętrostwo (gdy jądra pozostają w jamie brzusznej, zamiast zejść do worka mosznowego);
- zez;
- zaburzenia refrakcji (krótkowzroczność, astygmatyzm);
- jaskra/podwyższone ciśnienie śródgałkowe.
Minimalne kryteria sugerujące mcEDS
- Noworodki i małe dzieci: I kryterium większe i II kryterium większe
- Nastolatkowie i dorośli: I kryterium większe i III kryterium większe
EDS z przykurczami mięśniowymi jest związany z mutacjami genów CHST14 i DSE, powodującymi zmiany w białkach D4ST1 i DSE.
Mutacje innych genów wykazujących objawy kliniczne: PLOD1; FKBP14; ZNF469; PRDM5; B4GALT7; B3GALT6 i SLC39A13.
Brak mutacji w jednym lub więcej wyżej wymienionych genach nie wyklucza diagnozy. Przy braku mutacji w genie CHST14 lub DSE powinno się rozważyć inną możliwą diagnozę.
Sposób dziedziczenia: autosomalny recesywny.
Typ miopatyczny (mEDS, ang. myopathic EDS)
Kryteria większe:
I Kryterium: wrodzona hipotonia mięśniowa i/lub atrofia mięśni poprawiająca się wraz z wiekiem,
II Kryterium: przykurcze stawów proksymalnych (bliższych, jak kolana, biodra, łokcie),
III Kryterium: hipermobilność stawów dystalnych (dalszych).
Kryteria mniejsze:
- delikatna, ciastowata skóra,
- blizny atroficzne (przerostowe),
- opóźnienie rozwoju motorycznego,
- miopatia w obrazie histologicznym z biopsji mięśnia.
Minimalne kryteria sugerujące mEDS:
- I kryterium większe,
- Inne kryterium większe i/lub 3 kryteria mniejsze.
Miopatyczny typ EDS związany jest z heterozygotyczną lub bialleliczną mutacją w genie COL12A1 powodujący uszkodzenie kolagenu typu XII.
Mutacje innych genów u pacjentów wykazujących objawy kliniczne: COL6A1, COL6A2, COL6A3.
Brak mutacji w wyżej wymienionych genach nie wyklucza diagnozy mEDS. Przy braku mutacji w genie COL12A1 powinny zostać rozważone inne możliwe diagnozy, jak np. Wrodzona dystrofia mięśniowa Ullricha związana z kolagenem typu VI lub Miopatia Bethlema.
Sposób dziedziczenia: autosomalny dominujący lub autosomalny recesywny.
Typ przyzębowy (pEDS, ang. periodontal EDS)
Kryteria większe:
I Kryterium: poważne i trudne w leczeniu choroby przyzębia wcześnie się pojawiające (w dzieciństwie lub w wieku dojrzewania),
II Kryterium: odsłonięte szyjki zębowe z powodu braku przylegających dziąseł,
III Kryterium: narośla, zgrubienia na piszczeli (przednia strona podudzia),
IV Krewny pierwszego stopnia spełniający kryteria kliniczne w wywiadzie,
Kryteria mniejsze:
- łatwość siniaczenia,
- hipermobilność stawów, głównie dystalnych (dalszych),
- rozciągliwa i delikatna skóra z anormalnymi bliznami (szerokie lub atroficzne – zanikowe),
- nawracające infekcje,
- przepukliny,
- marfanoidalna budowa twarzoczaszki,
- akrogeria – starczy wygląd dłoni/stóp,
- widoczne/wydatne naczynia krwionośne.
Minimalne kryteria sugerujące pEDS:
- I kryterium większe lub II kryterium większe.
- 2 inne kryteria większe i jedno kryterium mniejsze.
Przyzębowy typ EDS jest związany z mutacjami genów: C1R lub C1S
Sposób dziedziczenia: autosomalny dominujący.
Typ XIV, nieokreślony
Stwierdzony jedynie u 4 osób z 3 niespokrewnionych ze sobą rodzin. Chorzy spełniali kliniczne kryteria cEDS, vEDS oraz aEDS, jednak badania genetyczne nie potwierdziły żadnego z nich. Objawy obejmują: wiotkość stawową, zwyrodnienia stawów, nadmierną ilość (jak akrogeria) i hiperelastyczność skóry, słabe gojenie się ran z atroficznym bliznowaceniem, osteoporozę, dyslokacje ramion, bioder, kolan i stawów skokowych; a także problemy gastryczne (pęknięcia jelit, zaburzenia perystaltyki, przepukliny), moczowo-płciowe (wnętrostwo); naczyniowe (wypadanie zastawki mitralnej, rozszerzenie opuszki aortalnej).
Kryteria diagnostyczne nie zostały jeszcze ustalone.
Typ XIV jest związany z bialleliczną mutacją genu AEBP1.
Sposób dziedziczenia: autosomalny recesywny.
Choroby współwystępujące w Zespole Ehlersa – Danlosa
Mięśniowo-szkieletowe
- częste podwichnięcia i zwichnięcia stawów;
- niestabilność kręgosłupa (w tym niestabilność szczytowo-potyliczna i szczytowo-obrotowa);
- skolioza, kifoskolioza (wyraźne zaznaczone w kEDS);
- wczesne zwyrodnienia stawów;
- osteopenia, osteoporoza (nawet u dzieci);
- niestabilność stawu skroniowo żuchwowego (TMJ);
- cieśnie: np. nadgarstka, podkolanowa, TOS;
- patologiczne napięcie mięśni;
- płaskostopie poprzeczne i podłużne;
- stopy/kolana końsko-szpotawe.
Gastroenterologiczne
- zespół jelita drażliwego;
- gastropareza;
- eozynofilowe zapalenie przełyku;
- liczne nietolerancje pokarmowe (w tym nietolerancja glutenu, laktozy, fruktozy, histaminy);
- refluks;
- zgaga;
- krwawienie do przewodu pokarmowego;
- zaparcia, biegunki;
- dysfagia;
- wgłobienia jelit;
- zaburzenia funkcji zwieraczy (np. żołądkowo-przełykowego);
- przepuklina przeponowa;
- opadanie narządów wewnętrznych;
- wypadanie narządów;
- pękanie narządów i dużych naczyń krwionośnych (głównie vEDS).
Sercowo-naczyniowe
- malformacje naczyniowe: tętniaki, rozwarstwienia naczyń, żylaki, zapadanie żył (w tym żył szyjnych i mózgowych);
- wypadanie zastawek serca;
- Posturalna Tachykardia Ortostatyczna;
- Nietolerancja Ortostatyczna;
- Hipotonia Ortostatyczna;
- omdlenia wazowagalne.
Neurologiczne
- dysautonomia;
- bóle głowy (w tym migrenowe, napięciowe i szyjnopochodne)
- malformacja Chiari'ego;
- obniżenie migdałków móżdżku (ang. cerebellar tonsillar ectopia);
- zakotwiczenie rdzenia kręgowego;
- jamistość rdzenia;
- zaburzenia propriocepcji;
- dystonia;
- chroniczny ból – może być uogólniony lub miejscowy;
- zespół bólu regionalnego;
- central sensitization;
- torbiele tarlowa;
- labilne ciśnienie śródczaszkowe (nadciśnienie śródczaszkowe z objawami niedociśnienia) – idiopatyczne nadciśnienie śródczaszkowe/pseudoguz mózgu (ang. pseudotumor cerebri), konsekwencją którego jest wyciek płynu mózgowo-rdzeniowego – przy czym w badaniach ciśnienie wychodzi w normie, diagnozę stawia się na podstawie poprawy lub pogorszenia objawów po punkcji lędźwiowej;
- chroniczne zmęczenie.
Immunologiczne
- Zespół Aktywacji Komórek Tucznych (MCAS, ang. Mast Cell Activation Syndrome);
- liczne alergie i/lub nietolerancje.
Choroby jamy ustnej i dziąseł
- łatwe uszkodzenia śluzówki jamy ustnej;
- parodontoza;
- hipoplazja szkliwa;
- złamania zębów.
Zaburzenia wzroku
- szybko postępujące wady wzroku (krótkowzroczność, dalekowzroczność);
- rozwarstwienie siatkówki;
- astygmatyzm.
Zaburzenia psychiatryczne
- zaburzenia snu;
- depresja;
- stany lękowe.
Stowarzyszenie Ehlers-Danlos Polska
Od 2017 r. działa w Polsce Stowarzyszenie chorych na Zespół Ehlersa-Danlosa.
Bibliografia
- Dominique P. Germain, Ehlers-Danlos syndrome type IV, „Orphanet Journal of Rare Diseases”, 2, 2007, s. 32, DOI: 10.1186/1750-1172-2-32, PMID: 17640391, PMCID: PMC1971255.
- Gernot Rassner, Dermatologia. Podręcznik i atlas, Wrocław: Urban & Partner, 1994, ISBN 83-85842-25-X.
- Vinay Kumar, Ramzi S. Cotran, Stanley L. Robbins, Patologia Robinsa, Wrocław: Urban & Partner, 2005, ISBN 0-7216-9274-5.
- Jerzy Stachura, Wenancjusz Domagała, Patologia znaczy słowo o chorobie, Kraków: Wydawnictwo PAU, 2003, ISBN 83-88857-65-7.
- Andrzej Szczeklik, Choroby wewnętrzne, Kraków: Medycyna Praktyczna, 2005, ISBN 83-7430-069-8.
- Fransiska Malfait i inni, The 2017 international classification of the Ehlers-Danlos syndromes, „American Journal of Medical Genetics Part C (Seminars in Medical Genetics)”, 175 (C), 2017, DOI: 10.1002/ajmg.c.31552, PMID: 28306229.
- Olga Haus, Nowa Klasyfikacja Zespołu Ehlersa-Danlosa, Prezentacja przedstawiona podczas I Bydgoskiego Spotkania Osób z Zespołem Ehlersa-Danlosa, 20 maja 2017 [dostęp 2019-08-10].
- Fraser C. Henderson, Neurological and spinal manifestations of the Ehlers-Danlos syndromes, „American Journal of Medical Genetics Part C (Seminars in Medical Genetics)”, 175 (1), 2017, s. 195-211, DOI: 10.1002/ajmg.c.31549, PMID: 28220607.
Linki zewnętrzne
- The Ehlers Danlos Society, www.ehlers-danlos.com [dostęp 2019-08-10] (ang.).
- Germaine L Defendi, Genetics of Ehlers-Danlos Syndrome, www.emedicine.com, 2 listopada 2017 [dostęp 2019-08-10] (ang.).