

Enzymy (z gr. ἔνζυμον, od ἔν en „w” i ζύμη dzýmē „zaczyn (za)kwas”) – wielkocząsteczkowe, w większości białkowe, katalizatory przyspieszające specyficzne reakcje chemiczne poprzez obniżenie ich energii aktywacji.
Niemal wszystkie reakcje chemiczne związane z funkcjonowaniem organizmów żywych (a także wirusów) wymagają współudziału enzymów, by osiągnąć wystarczającą wydajność. Enzymy są wysoce specyficzne wobec substratów i wobec tego dany enzym katalizuje zaledwie kilka reakcji spośród wielu możliwych dla danych substratów. W ten sposób enzymy determinują procesy metaboliczne i biochemiczne związane z funkcjonowaniem organizmów żywych.
Jak wszystkie katalizatory, enzymy obniżają energię aktywacji (Ea lub ΔG‡) reakcji chemicznej, przyspieszając w ten sposób przebieg reakcji (patrz: Struktury i mechanizmy działania). Większość reakcji enzymatycznych (tj. z udziałem enzymów) przebiega miliony razy szybciej niż ich niekatalizowane enzymatycznie odpowiedniki. Jednym z najszybciej działających znanych enzymów jest anhydraza węglanowa. Jedna cząsteczka tego enzymu potrafi w sprzyjających warunkach w jedną sekundę uwodnić od 104 do 106 cząsteczek dwutlenku węgla. Z kolei jedna cząsteczka jednego z najwolniejszych enzymów – lizozymu, katalizuje 1 akt elementarny co 2 sekundy. Jak wszystkie katalizatory, również enzymy nie zużywają się w trakcie przebiegu reakcji, a także nie wpływają na ich równowagę. Enzymy różnią się od zwykłych katalizatorów, przejawiając znacznie większą specyficzność substratową. Aktywność enzymatyczna może być zatrzymana lub obniżona przez inne cząsteczki – inhibitory. Wiele leków i trucizn jest inhibitorami enzymów. Z kolei aktywatory enzymatyczne to cząsteczki zwiększające aktywność enzymów. Ponadto aktywność enzymów zależy od parametrów fizykochemicznych środowiska reakcji, takich jak: temperatura, pH, siła jonowa, obecność niektórych jonów i innych.
Znane są także biokatalizatory niebiałkowe. Należą do nich rybozymy, cząsteczki RNA o własnościach katalitycznych oraz deoksyrybozymy (DNAzymy) – fragmenty DNA zdolne do katalizowania pewnych reakcji. Enzymy niebiałkowe charakteryzują się nieco innymi mechanizmami reakcji i mniejszą różnorodnością katalizowanych reakcji, jednak ich kinetyka i mechanika działania może być analizowana i klasyfikowana za pomocą tych samych metod, jakie są używane dla enzymów białkowych. Istnieją ponadto sztucznie stworzone cząsteczki, zwane sztucznymi enzymami, które przejawiają podobną do enzymatycznej aktywność katalityczną.
Liczne enzymy znalazły zastosowanie przemysłowe (patrz: Zastosowanie przemysłowe), m.in. w przemyśle spożywczym czy chemii leków. Wiele produktów używanych w gospodarstwach domowych zawiera enzymy w celu podniesienia wydajności ich działania, jak proszki do prania czy enzymatyczne wywabiacze do plam. Enzymy są także powszechnie używane we współczesnych naukach biologicznych i medycznych oraz w diagnostyce medycznej.
Badaniem enzymów i ich działania zajmuje się enzymologia.
Etymologia nazwy i historia odkrycia

Na przełomie XVIII i XIX wieku obserwowano już trawienie in vitro mięsa przez wydzieliny żołądka oraz rozkład skrobi do cukrów prostych przez ekstrakty roślinne lub ślinę, jakkolwiek mechanizmy stojące za tymi procesami nie były znane.
W XIX wieku, podczas studiów nad fermentacją cukrów do alkoholu przeprowadzaną przez drożdże, Louis Pasteur doszedł do wniosku, że reakcja ta jest ściśle powiązana z procesami życiowymi w drożdżach, którą nazwał „fermentem”. Podejrzewano, że może być ona aktywna tylko w żywych organizmach („nie ma fermentacji bez życia”). Pasteur odnotował, że „fermentacja alkoholowa jest procesem powiązanym z życiem i organizacją komórek drożdżowych, a nie ze śmiercią czy rozkładem komórek”.
W 1878 roku niemiecki fizjolog Wilhelm Kühne, by opisać te procesy, ukuł termin enzym, który pochodzi z greckiego ενζυμον i oznacza „w zaczynie”. Później słowo enzym było używane w odniesieniu do substancji nieożywionych, jak chociażby pepsyna, a słowo ferment w odniesieniu do aktywności chemicznej wykazywanej przez organizmy żywe. W języku polskim oba terminy były stosowane wymiennie, przy czym ten ostatni jest obecnie archaizmem.
W roku 1897 Eduard Buchner rozpoczął badania nad zdolnością ekstraktów drożdżowych do fermentacji cukrów przy nieobecności żywych komórek. Po szeregu eksperymentów przeprowadzonych na Uniwersytecie Humboldtów w Berlinie stwierdził on, że taka fermentacja jest możliwa. Odkryty enzym przeprowadzający fermentację sacharozy nazwał zymazą. W 1907 roku Eduard Buchner otrzymał Nagrodę Nobla w dziedzinie chemii „za swoje badania biochemiczne i odkrycie fermentacji bez udziału komórek”. Zgodnie z nazwą pierwszego enzymu, obecna nomenklatura ich nazewnictwa, przewiduje tworzenie nazwy od reakcji, którą przeprowadzają (patrz: Konwencja nazewnictwa). Zazwyczaj dodaje się przyrostek -aza do nazwy substratu przetwarzanego w reakcji (np. laktaza to enzym przetwarzający laktozę) lub do ogólnej nazwy reakcji (np. polimeraza DNA to enzym syntezujący polimery DNA).

Po udowodnieniu, że enzymy mogą funkcjonować poza komórką żywą, następnym krokiem było opisanie ich natury biochemicznej. Wielu badaczy zaobserwowało, że aktywność enzymatyczna była w jakiś sposób związana z białkami, ale kilku naukowców (w tym laureat Nagrody Nobla Richard Willstätter) dowodziło, że białka nie mogą być jedynymi „nośnikami” aktywności enzymatycznej i same z siebie nie są zdolne do przeprowadzania katalizy. Jednak w 1926 roku James Sumner udowodnił, że enzym ureaza to czyste białko i wyizolował je w formie krystalicznej. Powtórzył to także w 1937 roku dla katalazy. Ostateczny wniosek, że enzymy mogą być czystymi białkami, został potwierdzony przez Johna Howarda Northropa i Wendella Mereditha Stanleya, którzy pracowali nad enzymami trawiennymi – trypsyną i chymotrypsyną. Ta trójka naukowców została za swoje badania uhonorowana w 1946 roku Nagrodą Nobla w dziedzinie chemii.
Odkrycie, że enzymy mogą być wykrystalizowane pozwoliło na późniejsze badanie ich struktur metodami rentgenografii strukturalnej. Pierwszym enzymem, którego strukturę rozwiązano w ten sposób, był lizozym, enzym obecny m.in. w łzach, ślinie i białku jaja. Dokonała tego w 1965 roku grupa Davida Phillipsa. Uzyskanie trójwymiarowego modelu struktury lizozymu było początkiem biologii strukturalnej enzymów i zrozumienia mechanizmów ich działania na poziomie molekularnym.
W roku 1967 Carl Woese, Francis Crick i Leslie Orgel po raz pierwszy zasugerowali, że prócz białek, także RNA może przejawiać właściwości katalityczne. Aktywność katalityczną RNA odkrył Thomas Cech w roku 1980 przy badaniu splicingu RNA oraz, niezależnie, Sidney Altman podczas badań nad bakteryjną RNazą P. Katalityczne RNA zostały zaobserwowane w samowycinających się sekwencjach intronowych genów. W 1989 roku Thomas Cech i Sidney Altman zostali uhonorowani Nagrodą Nobla w dziedzinie chemii za swoje odkrycie „katalizujących właściwości RNA”. Termin rybozym, połączenie słów ryboza i enzym; po raz pierwszy został użyty w odniesieniu do tych cząsteczek przez Kelly Kruger i Thomasa Cecha w ich publikacji z 1982 roku.
Struktury i mechanizmy działania

Większość znanych enzymów to białka o zróżnicowanej wielkości, od kilkudziesięciu aminokwasów w łańcuchu i monomerycznej budowie (na przykład tautomeraza 4-oksokrotonianu (4-OT) zbudowana jest z 62 aminokwasów), do ponad 2500 w zwierzęcej syntazie kwasów tłuszczowych. Aktywność enzymów jest determinowana ich strukturą czwartorzędową (ułożeniem przestrzennym). Enzymy są zazwyczaj dużo większe od substratów, które przerabiają, ale z kolei zwykle tylko kilka kluczowych aminokwasów jest bezpośrednio zaangażowanych w katalizę. Region, który bezpośrednio wiąże się i oddziałuje z substratem oraz zawiera kluczowe do przebiegu reakcji reszty aminokwasowe, nazywany jest miejscem aktywnym (centrum aktywnym). Prócz niego enzymy mogą zawierać miejsca wiązania kofaktorów – niezbędnych do aktywności enzymatycznej lub jej regulacji (patrz: Kofaktory, grupy prostetyczne i koenzymy). Enzymy mogą także zawierać dodatkowe miejsca wiązania małych cząsteczek, na przykład pośrednich lub bezpośrednich produktów czy substratów szlaków metabolicznych obsługiwanych przez enzym, które związane mogą dodatkowo regulować aktywność enzymu na zasadzie sprzężenia zwrotnego.
Jak każde białko, enzymy są syntezowane jako długie łańcuchy aminokwasowe, które następnie zwijają się i przybierają odpowiednią strukturę przestrzenną. Indywidualne, zwinięte łańcuchy białkowe, mogą także asocjować w większe kompleksy. Takie enzymy nazywa się wtedy multimerycznymi (wielopodjednostkowymi). W przypadku asocjacji kilku takich samych peptydów (podjednostek) mówi się o homomerach (np. homodimer – kompleks złożony z dwóch jednakowych peptydów), a gdy asocjują różne jakościowo podjednostki, o heteromerach (np. heteropentamer – kompleks pięciu różnych łańcuchów peptydowych). Asocjacja podjednostek enzymów może być wymagana by dopełnić nawzajem swoje funkcje, by w ogóle móc katalizować reakcję biochemiczną, lub by obsługiwać wielokrotność tej samej reakcji czy cały ich szereg (odcinek szlaku metabolicznego).
Specyficzność
Enzymy charakteryzują się zwykle dużą specyficznością pod względem katalizowanej reakcji, jak i również konwertowanych substratów. Za wysoką specyficzność odpowiada kształt cząsteczki enzymu dopasowany do substratów geometrycznie, ale także pod względem oddziaływań hydrofobowo-hydrofilowych oraz elektrostatycznych. Enzymy wykazują także wysoki poziom stereospecyficzności, regioselektywności i chemoselektywności.
Niektóre z enzymów zaangażowanych w kopiowanie i ekspresję informacji genetycznej, poza bardzo wysoką specyficznością i precyzją, wykazują także zdolność działania „korekcyjnego” (ang. reading-proof activity). Przykładem może być polimeraza DNA I, która katalizuje syntezę nici DNA w czasie jej replikacji i naprawy. Polimeraza ta nie tylko jest wysoce precyzyjna, ale i zdolna do natychmiastowej poprawy ewentualnie zaistniałego błędu (źle wbudowanego nukleotydu). W rezultacie, dzięki tym dwóm właściwościom (precyzja syntezy i korekcja błędów), ryzyko popełnienia błędu przez ssacze polimerazy, który nie zostanie zauważony w procesie syntezy, wynosi mniej niż jeden na miliard wprowadzonych nukleotydów. Podobne mechanizmy korekcyjne posiadają polimerazy RNA, syntetazy aminoacylo tRNA i rybosomy.
Z kolei niektóre enzymy uczestniczące w produkcji metabolitów wtórnych charakteryzują się stosunkowo szerokim zakresem akceptacji różnych substratów. Sugeruje się, że odgrywają one bardzo ważną rolę w ewolucji nowych szlaków metabolicznych.
Model „klucza i zamka” (ang. „Lock and key” model)
W większości przypadków enzymy są niezwykle specyficzne wobec swoich substratów. Jak sugerował w roku 1894 Hermann Emil Fischer, zarówno enzym, jak i jego substraty są do siebie geometrycznie dopasowane w taki sposób, że idealnie pasują jeden do drugiego (jak „klucz i zamek”). Model ten wyjaśnia specyficzność enzymów, ale nie wyjaśnia w jaki sposób stabilizowany jest stan przejściowy podczas reakcji enzymatycznej.
Model „trzypunktowego dołączenia” (ang. Three-point interaction model)

W 1948 roku Alexander George Ogston, interpretując wyniki badań prowadzonych nad procesami metabolicznymi, w których wykorzystano izotopowe znakowanie substratów w reakcji enzymatycznej, zauważył, że do specyficznego związania substratu przez enzym wystarczą tylko trzy miejsca wiązania. Jeśli substrat może się przyłączyć do miejsca katalitycznego tylko z jednej strony i mogą ze sobą reagować tylko atomy i miejsca komplementarne, cząsteczka substratu może się przyłączyć tylko w jeden sposób. Oznacza to, że symetryczna cząsteczka substratu staje się poniekąd asymetryczna dla wypadku katalizy enzymatycznej. Przykładem jest kwas aminomalonowy wiążący się do modelowego, planarnego miejsca katalitycznego na enzymie. Kwas aminomalonowy zawiera dwie identyczne grupy karboksylowe, które stają się przestrzennie różne po trzypunktowym związaniu cząsteczki. Zatem jeśli reakcja obejmuje zawsze jedną grupę karboksylową w konkretnym miejscu, wówczas reagować będzie zawsze ta sama grupa karboksylowa. Jednakże model ten jest wyraźnie jakościowy, dostarczając tylko ograniczonego wyjaśnienia ilościowych i energicznych parametrów przebiegu stereospecyficznych procesów. Rozszerzenie modelu dla większej ilości punktów wiązania, co zapewnia większą precyzję w doborze substratu, opisali Ran Kafri i Doron Lancet w 2004 roku.
Model indukowanego dopasowania (ang. Induced fit model)
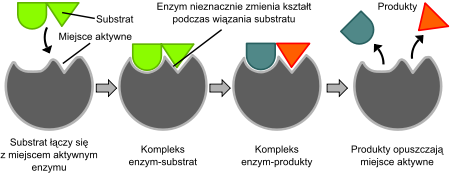
W 1958 roku Daniel Koshland zaproponował modyfikację modelu „klucza i zamka”. Ponieważ enzymy są zwykle dość elastyczne strukturalnie, ich centrum aktywne podlega ciągłym rearanżacjom przestrzennym podczas oddziaływania z substratami. W rezultacie, substrat nie tyle wiąże się do niezmiennego strukturalnie miejsca aktywnego, ale grupy boczne aminokwasów je tworzące podlegają rearanżacjom przestrzennym, ściśle dopasowując swe pozycje do wiązanego substratu, co dopiero umożliwia przeprowadzenie katalizy. W niektórych wypadkach, jak np. glikozydazy, także cząsteczki substratu podlegają lekkim zmianom strukturalnym po wejściu do centrum aktywnego, wzmacniając w ten sposób dopasowanie. Centrum aktywne kontynuuje rearanżację, aż osiągnięte zostanie końcowe stadium dopasowania z substratem.
Mechanizmy
Enzymy mogą na kilka różnych sposobów zmniejszać swobodną energię aktywacji Gibbsa (ΔG‡):
- Obniżanie energii aktywacji przez tworzenie środowiska, w którym następuje stabilizacja stanu przejściowego (np. zniekształcenie cząsteczki substratu – dzieje się to przez utrwalenie konformacji stanu przejściowego pomiędzy substratem a produktem oraz zniekształcenie przez enzym wiązań w cząsteczce substratu, dzięki czemu zmniejsza się suma energii wymaganej do zakończenia przejścia).
- Obniżanie energii stanu przejściowego przez likwidację niekorzystnych energetycznie oddziaływań ze środowiskiem, i ich zamianę na korzystne energetycznie oddziaływania z centrum aktywnym (np. oddziaływania elektrostatyczne ładunków o przeciwnych znakach).
- Wykorzystanie alternatywnego szlaku przejścia, np. tymczasowa reakcja substratu do pośredniego kompleksu enzym-substrat (ES), która nie byłaby możliwa w nieobecności enzymu.
- Zmniejszanie entropii reakcji poprzez usytuowanie dostarczanych razem substratów w poprawnej orientacji, niezbędnej do zajścia reakcji. Rozpatrując energetykę reakcji enzymatycznej pod kątem jedynie zmiany entalpii (ΔH‡), efekt ten jest zwykle pomijany. Wiąże się to z faktem, że ma on miejsce tylko dla stanu podstawowego reakcji (substraty), a jego wpływ na właściwą katalizę jest znikomo mały.
Stabilizacja stanu przejściowego
Enzymy w obsługiwanych reakcjach obniżają energię aktywacji (ΔG‡) poprzez stabilizację stanu przejściowego. Jest to możliwe poprzez niezwykle efektywne wykorzystanie efektów oddziaływań elektrostatycznych pomiędzy cząsteczkami w stanie przejściowym a otaczającym je środowiskiem. W reakcji niekatalizowanej kompleks stanu przejściowego (S*) oddziałuje z otaczającą wodą w sposób mało uporządkowany, co zmusza system do pokonania dużej bariery energetycznej (energia aktywacji). Enzym, zapewniając ściśle dopasowaną „kieszeń” centrum aktywnego, jest w stanie zagwarantować optymalne energetycznie oddziaływanie kompleksu stanu przejściowego (ES*) z otaczającym je środowiskiem – na przykład zapewniając mu kontakt z resztami aminokwasowymi centrum aktywnego dokładnie pasującymi elektrostatycznie do rozkładu ładunków na powierzchni kompleksu stanu przejściowego. Dodatkowo, samo zbliżenie do siebie substratów, a także zapewnienie odpowiednich donorów/akceptorów elektronów zachodzącej reakcji, obniża koszty energetyczne jej przebiegu, co nie ma miejsca dla reakcji zachodzącej spontanicznie w wodzie.
Dynamika enzymów
Nowe badania pozwoliły głębiej poznać związek między dynamiką enzymów a mechanizmem ich katalizy. Dynamika wewnętrzna enzymów jest opisywana jako ruch wewnętrznych fragmentów (np. aminokwasów, grup aminokwasów, regionów pętli, alfa-helis, obszarów beta-kartek lub nawet całych domen) tych biocząsteczek, który może zajść w szerokim przedziale czasowym, wahającym się od kilku femtosekund do sekundy. Pozostałe obszary łańcuchów białkowych tworzą rusztowanie dla funkcjonalnych obszarów cząsteczki oraz umożliwiają ich dynamiczne ruchy, wspomagając w ten sposób katalizę. Białkowe ruchy są istotne w obrębie cząsteczki wielu enzymów, ale czy będą to małe i szybkie ruchy czy też duże i powolne zmiany konformacyjne, zależne jest od typu katalizowanej reakcji, w którą zaangażowany jest konkretny enzym. Dynamika enzymów prawdopodobnie nie ma jednak wpływu na ogólną szybkość reakcji enzymatycznej.
Modulacja allosteryczna
Niektóre enzymy, to enzymy allosteryczne, zmieniające swoją konformację w odpowiedzi na związanie efektora (inhibitora lub aktywatora). Modulacja taka może być bezpośrednia; gdy efektor sam wiąże się z enzymem, lub pośrednia; gdy efektor wiąże się z innym białkiem, lub podjednostką białka, która oddziałuje z enzymem, zmieniając jego aktywność katalityczną.
Kofaktory, grupy prostetyczne i koenzymy

Wiele enzymów potrzebuje dodatkowych składników do uaktywnienia czy osiągnięcia pełnej aktywności. Takie niebiałkowe, dodatkowe składniki enzymów, nazywane są kofaktorami. Enzym bez swojego kofaktora, czyli sam jego białkowy składnik, to apoenzym, natomiast wraz z kofaktorem, katalitycznie aktywny enzym nazywany jest holoenzymem (sam termin enzym domyślnie oznacza właśnie jego kompletną, aktywną cząsteczkę, czyli holoenzym).
Kofaktory można podzielić na dwie szerokie grupy. Pierwszą z nich stanowią grupy prostetyczne, czyli kofaktory silnie, często kowalencyjnie, związane przez enzym przez cały czas jego istnienia. Zwykle są to cząsteczki nieorganiczne i jony metali (np. centra żelazowo-siarkowe, jony cynku – Zn2+, enzymy z metalami jako grupami prostetycznymi zwane są metaloenzymami), ale także małe cząsteczki organiczne (np. flawiny i hem). Z kolei koenzymy to małe, niebiałkowe cząsteczki organiczne, wiążące się z enzymami tylko na czas reakcji, i przenoszące grupy chemiczne pomiędzy poszczególnymi reakcjami. Koenzymy mogą być traktowane jako kosubstraty, ponieważ są wiązane i uwalniane z enzymów jak substraty i produkty oraz biorą bezpośredni udział w reakcji.
Niektóre źródła nazwę kofaktor przypisują cząsteczkom nieorganicznym, podczas gdy organiczne zwane są koenzymami.
Grupy prostetyczne
Silnie związane z enzymem grupy prostetyczne występują zwykle w jego miejscu aktywnym i są bezpośrednio zaangażowane w katalizę oraz nie opuszczają miejsca aktywnego podczas reakcji. Na przykład flawinowe i hemowe kofaktory biorą udział w reakcjach redoks.
Większość kofaktorów nie jest kowalencyjnie powiązana z enzymem. Jednakże organiczne grupy prostetyczne mogą się wiązać kowalencyjnie np. pirofosforan tiaminy w dehydrogenazie pirogronianowej.
Koenzymy
Koenzymy są małymi cząsteczkami organicznymi transportującymi grupy chemiczne pomiędzy różnymi reakcjami (substratami). Na przykład koenzymy nikotynamidowe, NAD+ lub NADP+, uczestniczą w transporcie elektronów i protonów, grupy acetylowe przenoszone są przez koenzym A, grupy: formylowe, metylowe lub metylenowe przenoszone są z udziałem kwasu foliowego, grupa metylowa może też być przenoszona przez S-adenozylometioninę. Niektóre z koenzymów, takich jak: ryboflawina, tiamina i kwas foliowy, są witaminami.
Ponieważ koenzymy podlegają zmianom chemicznym w wyniku działania enzymów, można je postrzegać jako specjalną klasę substratów (kosubstratów), czy też substratów wtórnych, wspólnych dla wielu różnych enzymów.
Stężenie koenzymów wewnątrz komórki jest utrzymywane na stałym poziomie dzięki ich ciągłej regeneracji na różnych szlakach biochemicznych. Np. NADPH jest regenerowany na szlaku pentozofosforanowym.
Termodynamika

Tak jak wszystkie katalizatory enzymy nie zmieniają stanu równowagi reakcji chemicznej, a jedynie przyspieszają jego ustalenie. Zazwyczaj w obecności enzymu reakcja zachodzi w kierunku takim samym, w jakim by zachodziła spontanicznie, jedynie wzrasta jej szybkość. Jednak nieobecność enzymu oznacza, że najbardziej wydajnie (najszybciej) będzie zachodzić reakcja najbardziej faworyzowana energetycznie (o najniższej energii S*) i nie zawsze oznacza to, że z grupy możliwych reakcji jest to ta sama, która byłaby katalizowana przez enzym. Enzym może uczynić reakcję bardziej faworyzowaną, która w normalnych warunkach nie byłaby energetycznie optymalna.
Jest to termodynamicznie możliwe, gdyż enzymy sprzęgają reakcje niekoniecznie wydajne energetycznie (takie, które spontanicznie zachodziłyby niezwykle wolno) z reakcjami wybitnie energetycznie korzystnymi, tak że taka para reakcji, w sumie, jest energetycznie faworyzowana i korzystna. Przykładem może być sprzężenie reakcji hydrolizy ATP z innymi licznymi reakcjami chemicznymi, wymagającymi do zajścia dużej ilości energii.
Enzymy zwiększają szybkość reakcji zachodzącej w obu kierunkach. Na przykład anhydraza węglanowa katalizuje swoją reakcję dwukierunkowo w zależności od stosunku stężenia substratów i produktów.


Jednak, gdy w danych warunkach szybkość reakcji zachodzącej w konkretną stronę jest bardzo duża (równowaga mocno przesunięta w jedną ze stron), w praktyce oznacza to katalizę nieodwracalną i reakcja zachodzi tylko w tym jednym kierunku.
Kinetyka

Kinetyka enzymów opisuje mechanizmy wiązania substratów przez enzymy oraz ich przekształcania w produkty. Dane wykorzystywane do analizy kinetycznej pochodzą z reakcji enzymatycznych przeprowadzonych w kontrolowanych warunkach umożliwiających śledzenie zmieniających się parametrów w czasie.
W 1902 roku Victor Henri zaproponował kwantytatywną teorię kinetyki enzymów, ale wyniki jego badań okazały się nieprzydatne, ponieważ zaniedbał on wpływ stężenia jonów wodorowych (pH). Dopiero kilka lat później, w 1909 roku, Peter Lauritz Sørensen zdefiniował logarytmiczną skalę pH oraz zaproponował koncepcję roztworów buforowych. Niemiecki chemik Leonor Michaelis i jego kanadyjska współpracowniczka odbywająca staż podoktorski Maud Leonora Menten, powtórzyli wtedy eksperymenty, które przeprowadzał Henri i zaproponowali prosty model kinetyki aktywności enzymatycznej znany jako kinetyka Henri-Michaelis-Menten (znany również jako kinetyka Michaelis-Menten). Ich dzieło rozwinęli później G. E. Briggs i J.B.S. Haldane, których równania kinetyki aktywności enzymatycznej są do dziś używane w niezmienionej formie.
Głównym założeniem jakie podał Henri, był dwuetapowy przebieg reakcji katalizowanej przez enzymy. W pierwszym etapie substraty wiążą się odwracalnie z enzymem, tworząc ze stałą szybkości k1, kompleks enzym-substrat (ES), nazywany czasem kompleksem Michaelisa. W następnym etapie, kompleks ten może się rozpaść na dwa różne sposoby. Może dysocjować do E i S ze stałą szybkości k−1 lub może dojść do chemicznej zmiany substratów i uwolnienia produktów ze stałą szybkości k2, przy czym zakłada się, że produkt reakcji nie może ulec powrotnemu przekształceniu w wyjściowy substrat. Wyrażenie wiążące szybkość katalizy ze stężeniem substratu i enzymu oraz z szybkościami poszczególnych etapów reakcji nazywamy równaniem Michaelisa-Menten:

![{\displaystyle V_{0}=V_{max}{\frac {[S]}{[S]+K_{m}}},}](https://wikimedia.org/api/rest_v1/media/math/render/svg/1981f1423f58eebc2f9158c521e3bc6102449323)
gdzie:
- V0 – szybkość początkowa,
- S – stężenie substratu,
- Km – stała Michaelisa.
Enzymy mogą katalizować powyżej kilku milionów reakcji na sekundę, gdzie te same reakcje bez ich obecności mają czasy połowicznej przemiany substratu, tzn. czas w jakim pod nieobecność enzymu, połowa dostępnego substratu zostanie zużyta, kilkanaście rzędów wielkości dłuższy (21 rzędów wielkości to najwyższe znane przyspieszenie reakcji enzymatycznej, z tryliona lat do dziesiątek milisekund). Przykładem jest reakcja katalizowana przez dekarboksylazę orotydyno 5'-monofosforanu. Czas połowicznej przemiany reakcji nieenzymatycznej wynosi 78 mln lat, a czas połowicznej przemiany tej samej reakcji, ale z udziałem dekarboksylazy orotydyno 5'-monofosforanu, wynosi 25 milisekund. Bezpośrednia szybkość z jaką działają enzymy zależy od stężenia substratów w roztworze (dodatkowo szybkość enzymów zależy od ogólnych parametrów fizykochemicznych środowiska, tak jak w przypadku aktywności każdego białka, patrz: Aktywność a parametry środowiska). By znaleźć maksymalną szybkość reakcji katalizowanej enzymatycznie, określa się ilość tworzonego produktu w funkcji czasu, przy stopniowym zwiększaniu stężenia substratu, ale jednoczesnym zachowaniu innych warunków, do momentu, w którym dalszy wzrost jego stężenia nie powoduje dalszego przyśpieszania reakcji. Szybkość początkowa w takim eksperymencie (V0), rośnie do wartości maksymalnej (Vmax) i nie przekracza jej mimo dalszego wzrostu stężenia substratu. W chwili osiągnięcia stanu równowagi (reakcji katalizowanej ze stałą szybkości k2 i reakcji do niej przeciwnej zachodzącej ze stałą szybkości k−2), nie obserwujemy zmiany netto stężenia substratów i produktów. Zależność szybkości reakcji od stężenia substratu dla prostej reakcji według kinetyki Michaelisa-Menten, przy założeniu, że kompleks ES jest koniecznym etapem pośrednim procesu katalitycznego, przedstawia krzywa Michaelisa-Menten. Wysycanie enzymu substratem (zbliżanie się do szybkości maksymalnej) oznacza, że maleje liczba wolnych cząsteczek enzymu, gdyż rośnie ilość tych związanych w kompleksie z substratem (ES). Osiągana asymptotycznie maksymalna szybkość (Vmax) reakcji enzymatycznej, oznacza, że praktycznie wszystkie miejsca aktywne enzymu (wolne enzymy) zostają wysycone substratem, a wówczas suma ilości (stężenie) kompleksów ES jest miarą ilości samego enzymu. Niemniej jednak, Vmax jest tylko jedną stałą kinetyczną opisującą enzym. Inną jest ilość substratu (stężenie) potrzebne do osiągnięcia ustalonej szybkości reakcji. Zwykle używa się do tego celu stałej Michaelisa (Km), która jest takim stężeniem substratu przy którym szybkość reakcji osiąga połowę swojej maksymalnej wartości. Każdy enzym ma swoją charakterystyczną wartość Km dla danego substratu, która charakteryzuje również siłę wiązania substratu w miejscu aktywnym enzymu. Inną użyteczną stałą jest szybkość katalizy (kkat), która jest liczbą obrotów jednego miejsca aktywnego enzymu w czasie jednej sekundy (liczba reakcji przeprowadzana przez jedno miejsce aktywne w czasie jednej sekundy).
Innymi jednostkami używanymi do opisu aktywności enzymów są: jednostka enzymu (U), czyli taka ilość enzymu, która katalizuje przemianę 1 μmola substratu w czasie 1 minuty w temperaturze 30 °C i określonym pH oraz katal (kat), czyli ilość enzymu katalizująca przemianę 1 mola substratu w czasie 1 sekundy. Dodatkowo dla niektórych enzymów używa się jednostek umownych, definiowanych przez zastosowany pomiar aktywności. Na przykład przyrost absorbancji światła o danej długości fali w danej jednostce czasu, gdy wynikiem reakcji enzymatycznej jest barwny produkt. W preparatyce biochemicznej dodatkowo używa się aktywności właściwej, która określa liczbę jednostek enzymu (lub jednostek umownych aktywności) przypadających na 1 mg białka (w danym preparacie).
W sytuacji, gdy stężenie substratu znacznie przewyższa Km, szybkość katalizy jest równa kkat, liczbie obrotów. Jednak w warunkach fizjologicznych, większość enzymów nie jest wysycona substratem, zatem stosunek [S]/Km mieści się w granicach od 0,01 do 1,0. Gdy stężenie substratu ([S]) jest dużo mniejsze od Km ([S]<<Km), wówczas szybkość katalizowanej reakcji jest znacznie mniejsza od kkat, ponieważ większość miejsc aktywnych nie jest zajęta. Do scharakteryzowania kinetyki enzymów w tych warunkach służy równanie V0=kkat/Km[E][S], powstałe z połączenia dwóch innych równań: V0=k2 [ES] oraz [ES]=[E][S] / Km. W przedstawionej sytuacji, gdy [S]<<Km to stężenie wolnego enzymu jest niemal równe całkowitemu stężeniu enzymu. W tych warunkach stosunek kkat/Km jest stałą szybkości oddziaływania S i E i może być użyty jako miara wydajności katalitycznej. Ponieważ stosunek kkat/Km odzwierciedla zarówno powinowactwo chemiczne, jak i zdolność katalityczną, jest używany do porównywania różnych enzymów względem siebie lub preferencji jednego enzymu do różnych substratów. W sytuacji, gdy szybkość tworzenia produktu (kkat) jest znacznie szybsza niż szybkość dysocjacji kompleksu ES, wartość kkat/Km zbliża się do wartości stałej tworzenia kompleksu ES. Z tego wynika, że górna granica wartości kkat/Km jest ograniczana przez szybkość tworzenia kompleksu ES, która nie może być większa niż częstość z jaka spotykają się enzym i jego substrat na skutek dyfuzji. Częstość ta mieści się w granicach od 108 do 109 (M−1 s−1) i jest to jednocześnie górna granica kkat/Km. W tym punkcie, każde zderzenie enzymu z jego substratem będzie powodowało katalizę i szybkość formowania produktu nie jest limitowana przez szybkość reakcji, ale przez szybkość dyfuzji. Enzymy posiadające tę własność nazywamy katalitycznie perfekcyjnymi lub kinetycznie perfekcyjnymi. Przykładami takich enzymów są: izomeraza triozofosforanowa, anhydraza węglanowa, esteraza acetylocholinowa, katalaza, fumaraza, β-laktamaza i dysmutaza ponadtlenkowa.
Kinetyka niektórych enzymów charakteryzuje się szybkością większą od teoretycznej szybkości dyfuzji. Jest to możliwe, gdyż wiązanie substratów przez enzym (krok limitowany przez szybkość dyfuzji) i tworzenie kompleksu ES są wspomagane dodatkowymi efektami niezależnymi od samej dyfuzji. Niektóre białka enzymatyczne aktywnie przeorientowują substraty za pomocą pól elektrycznych generowanych przez cząsteczki (reszty) dipolowe. Inne opierają się na kwantowym zjawisku tunelowym, w którym proton lub elektron może pokonać barierę potencjału o energii większej niż jego energia, nie przez „przeskakiwanie nad barierą”, a przez „przebijanie się przez nią”, chociaż dla protonu ten model wciąż jest przedmiotem dyskusji. Jakkolwiek zjawisko tunelowania zostało zaobserwowane w reakcji utleniania tryptaminy przez dehydrogenazę amin aromatycznych (ang. aromatic amine dehydrogenase, AADH).
Aktywność a parametry środowiska
Ogólna aktywność enzymów, podobnie jak wszystkich białek, jest zależna od parametrów fizykochemicznych środowiska: temperatury, pH, siły jonowej i innych. Maksimum aktywności enzymu leży w pewnym optymalnym zakresie danego parametru środowiskowego. W zależności od enzymu, położenie optimum może być różne, a jego zakres szerszy lub węższy. Także ogólny kształt wykresu zależności aktywności danego enzymu od parametru środowiska jest różny dla różnych enzymów, w zależności od ich pochodzenia, budowy itp. Dla czynników środowiska bezpośrednio wpływających na strukturę drugorzędowa białka (i wyższe organizacje struktury), jak temperatura czy pH, charakterystyczny jest gwałtowny spadek aktywności enzymów poza optimum (lub po przekroczeniu go, w przypadku temperatury), związany z denaturacją enzymów. W zależności od enzymu, taka denaturacja może być odwracalna lub nie. Parametry środowiska mogą stanowić podstawę kontroli aktywności enzymów (patrz: Kontrola aktywności).
Temperatura

Szybkość reakcji enzymatycznych wzrasta wraz ze wzrostem temperatury. Wiąże się to ze wzrostem energii kinetycznej cząstek i większą częstotliwością ich zderzeń. Wzrost aktywności enzymatycznej w zależności od temperatury opisuje współczynnik temperaturowy Q10, określający jak zmienia się szybkość reakcji przy wzroście temperatury o 10 °C:

Wpływ temperatury na aktywność enzymów nie jest prostą zależnością. Aktywność rośnie wraz ze wzrostem temperatury, jednakże tylko w takim zakresie temperatury, w którym enzym pozostaje stabilny. Po przekroczeniu temperatury krytycznej, następuje denaturacja termiczna enzymów, w wyniku czego aktywność gwałtownie spada. Przeciętnie szybkość reakcji enzymatycznych wzrasta dwukrotnie przy wzroście o każde 10 °C w zakresie temperatur niedenaturujących struktury enzymu (Q10=2). Zatem także parametr Q10 ma zastosowanie tylko w niedenaturującym zakresie temperatur i jest on charakterystyczny dla danego enzymu, i zależny od energii aktywacji katalizowanej reakcji. W temperaturze optymalnej aktywność enzymu jest największa. Obserwowana temperatura optymalna jest wypadkową dwóch procesów: wzrostu szybkości reakcji związanego ze wzrostem energii kinetycznej oraz wzrostu szybkości denaturacji termicznej enzymu powyżej krytycznej temperatury. Gdy drugi składnik zaczyna przeważać, następuje spadek aktywności. Większość enzymów ma optimum temperaturowe w zakresie 30–45 °C i nieodwracalnie denaturuje oraz traci aktywność w temperaturach wyższych niż 60 °C. Jednak w przypadku organizmów termofilnych (np. bakterii ze źródeł termalnych) ich enzymy zachowują aktywność i osiągają maksymalną szybkość w podwyższonych temperaturach.
Większość enzymów ulega powolnej denaturacji nawet w temperaturach optymalnych i niższych niż krytyczna. Zależy to od natury samego enzymu, stopnia jego oczyszczenia (w preparatach), a także od pH, siły jonowej i pozostałych parametrów. Najwyższa temperatura, w której jeszcze nie zachodzi termiczna dezaktywacja enzymu w danych warunkach określa tak zwaną termostabilność enzymu.
pH
Większość enzymów ma także swoje optymalne pH działania. Optimum pH, obok optimum temperaturowego, to drugi pod względem znaczenia parametr środowiska charakteryzujący aktywność enzymów. Wpływ pH na aktywność enzymów wiąże się z faktem, że enzymy jako białka posiadają wiele aminokwasów ulegających jonizacji, a aminokwasy centrum aktywnego często mogą pełnić swoją rolę tylko w określonym stanie jonizacji. Ponadto na aktywność enzymów, wpływ ma także jonizacja samych substratów oraz kompleksów ES. Również oddziaływania enzym-substrat mogą mieć charakter jonowy, zależny od jonizacji. Przykładem takiego zjawiska jest proces hydrolizy przeprowadzany przez pepsynę. Punkt izoelektryczny (pI) tego enzymu wynosi 1, natomiast optimum jego działania to około 2,0–2,5. W tym odczynie cząsteczka pepsyny, bogata w aminokwasy dikarboksylowe, jest jeszcze ujemnie naładowanym anionem, podczas gdy większość białek ma już przy tym pH ładunek dodatni. Od stopnia jonizacji pewnych grup zależy także ogólna konformacja cząsteczki enzymu, zapewniająca mu pełną aktywność, a w za kwaśnym czy zbyt zasadowym środowisku enzym ulegnie denaturacji.
Wykres zależności aktywności enzymu od pH ma najczęściej kształt dzwonowaty, czyli enzym ma największą aktywność w swoim optymalnym pH i aktywność ta spada wraz z oddalaniem się od tego punktu (np. trypsyna). Niektóre jednak enzymy mają właściwie stała aktywność w szerokim zakresie pH (np. papaina) lub są wrażliwe tylko na niskie pH, zachowując aktywność w wysokim (jak esteraza cholinowa) lub na odwrót. Zależność aktywności od pH bada się w stałej temperaturze przy maksymalnym wysyceniu enzymu substratem.
Inhibicja


Aktywność wielu enzymów może być hamowana przez różne typy inhibitorów. Inhibicja taka może być odwracalna lub nieodwracalna. Ostatnia ma miejsce wtedy, gdy cząsteczki inhibitora wiążą się z enzymem trwale (np. kowalencyjnie), co doprowadza do sytuacji zablokowania aktywności danej cząsteczki enzymu na stałe.
Typy inhibicji
Inhibicja kompetycyjna
W inhibicji kompetycyjnej inhibitor i substrat współzawodniczą o miejsce aktywne cząsteczki enzymu. Związanie przez enzym cząsteczki inhibitora uniemożliwia zatem związanie substratów (i vice versa) i kompleks enzym-inhibitor (EI) jest enzymatycznie nieaktywny. Zazwyczaj inhibitor kompetycyjny jest strukturalnie bardzo podobny (lub ma podobny motyw bezpośrednio wiążący się do centrum aktywnego) do prawdziwego substratu dla określonego enzymu. Na przykład metotreksat jest inhibitorem kompetycyjnym dla enzymu reduktazy dihydrofolianu, który katalizuje redukcję dihydrofolianu do tetrahydrofolianu i obie substancje są strukturalnie bardzo zbliżone. Przyłączenie inhibitora uniemożliwia związanie substratu i odwrotnie, dzięki czemu poprzez regulację stężenia inhibitora możliwa jest kontrola szybkości reakcji. Stałą dysocjacji inhibitora kompetycyjnego można opisać równaniem:
![{\displaystyle K_{i}={\frac {[E][I]}{[EI]}}.}](https://wikimedia.org/api/rest_v1/media/math/render/svg/989d49e0d3414bd12a3c7823c8aefef91d6855e9)
W inhibicji kompetycyjnej maksymalna szybkość reakcji (Vmax) nie zmienia się i może być osiągnięta poprzez zwiększenie stężenia substratów, które przezwycięży inhibicję. Zmianie natomiast ulega pozorna wartość Km, nowa wartość Km, zwana Kmapp (pozorna stała Michaelisa) jest równa:
![{\displaystyle K_{m}^{app}=K_{m}\left(1+{\frac {[I]}{K_{i}}}\right),}](https://wikimedia.org/api/rest_v1/media/math/render/svg/2c3c44b553e9cec3a2f81d477ef0a793dc8b4e0c)
gdzie:
- – stężenie inhibitora,
- – stała dysocjacji dla kompleksu enzym-inhibitor.
![{\displaystyle [I]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5962300a54e8ce8b5761dac9a5fbbca450c2ce0f)

Zatem wzrost [I] pociąga za sobą wzrost

Inhibicja akompetycyjna
W inhibicji akompetycyjnej inhibitor nie może się wiązać do wolnego enzymu, a jedynie do kompleksu enzym-substrat (ES), tworząc kompleks enzym-inhibitor-substrat (EIS). Ponieważ w takiej sytuacji zmniejszone jest stężenie kompleksu ES, zwiększa to pozorne powinowactwo enzymu do substratu, zatem wartość Km jest mniejsza. Utworzony kompleks EIS jest enzymatycznie nieaktywny i reakcja nie może być kontynuowana, dopóki miejsce aktywne enzymu nie zostanie zwolnione, co obniża Vmax w porównaniu do reakcji nieinhibowanej. Ten typ inhibicji jest dość rzadki i może dotyczyć niektórych enzymów multimerycznych (wielopodjednostkowych).
Inhibicja niekompetycyjna
Inhibitory niekompetycyjne mogą się wiązać do wolnego enzymu, jednak nigdy do jego miejsca aktywnego, wobec tego nie konkurują z substratami, które także mogą się przyłączyć do powstałego kompleksu. Oba możliwe kompleksy, enzym-inhibitor (EI) i enzym-inhibitor-substrat (EIS) są enzymatycznie nieaktywne. Ponieważ wiązanie inhibitora jest całkowicie niezależne od substratu, co oznacza, że większe stężenie substratu nie wpływa na obniżenie oddziaływań z inhibitorem (w przeciwieństwie do inhibicji kompetycyjnej), zatem zmienia się tylko wartość Vmax a wartość Km pozostaje stała.
Inhibicja mieszana
Ten typ inhibicji przypomina inhibicję niekompetycyjną, z wyjątkiem występowania dodatkowo kompleksu enzym-inhibitor-substrat (EIS), mającego znikomą aktywność enzymatyczną. W inhibicji mieszanej obie wartości opisujące reakcję enzymatyczną: Km i Vmax, zmieniają się.
Inhibicja nieodwracalna
Inhibitory nieodwracalne reagują z enzymem, wiążąc się kowalencyjnie, a zatem nieodwracalnie, do jego łańcuchów białkowych. Taka inhibicja trwale unieczynnia daną cząsteczkę enzymu. Do inhibitorów tego typu należy eflornityna, lek stosowany w pasożytniczej chorobie – śpiączce afrykańskiej. Również penicylina i aspiryna mają podobny mechanizm działania. Inhibitory te wstępnie, jeszcze nie kowalencyjnie, wiążą się z miejscami aktywnymi enzymów, po czym dopiero aktywność enzymu przekształca je w reaktywne formy, które wiążą się nieodwracalnie z jedną lub więcej resztami aminokwasowymi miejsca aktywnego enzymu, blokując je.
Regulacja aktywności enzymów przez inhibitory
W wielu reakcjach inhibitory biorą udział w mechanizmie regulacji aktywności enzymatycznej na drodze sprzężenia zwrotnego. Jeśli enzym produkuje jedną substancję ponad potrzeby komórki, to ta substancja może stać się inhibitorem dla tego enzymu, co zmniejsza lub całkowicie hamuje aktywność enzymu, co z kolei zmniejsza stężenie produktu. Taka regulacja jest formą ujemnego sprzężenia zwrotnego. Enzymy, które poddane są tego typu regulacji, to często układy wielopodjednostkowe, posiadające kilka miejsc aktywnych dla substancji regulatorowych. Dla takich enzymów, wykres prędkości katalizowanej reakcji w zależności od stężenia substratu, nie ma przebiegu hiperbolicznego, ale sigmoidalny (kształt litery S).
Kontrola aktywności
Istnieje kilka głównych sposobów kontroli aktywności enzymów w układach biologicznych:
- Sama produkcja enzymu na etapach translacji, jak i wcześniejszej transkrypcji, podlega ścisłej kontroli i może być zwiększana (uruchamiana) lub zmniejszana (zatrzymana) przez komórkę w odpowiedzi na zmiany w środowisku zewnętrznym komórki, tak samo jak kontrolowana jest w ten sposób produkcja każdego innego białka.
- Sortowanie i rozdzielanie enzymów do docelowych kompartmentów (także poza komórkę) i utrzymywanie ich tam, jest sposobem na kontrolę ich aktywności w przestrzeni. Enzymy działają wtedy na szlakach metabolicznych umiejscowionych w różnych przedziałach komórkowych, nie interferują z innymi szlakami, a ich aktywność może być także szybko zmieniona poprzez globalną zmianę charakterystyki środowiska kompartmentu w którym się znajdują (np. obniżenie pH w lizosomie). Różne kompleksy enzymatyczne zawarte w różnych przedziałach komórkowych, a co za tym idzie podział syntezy na etapy, jak również oddzielenie syntezy od rozkładu, umożliwia dokładną kontrolę szlaku metabolicznego na każdym etapie przez różne czynniki.
- Aktywność enzymów może być regulowana przez specjalne cząsteczki – inhibitory (patrz: Inhibicja) i aktywatory. Jest to najbardziej bezpośredni sposób modulacji ich aktywności.
- Enzymy mogą być regulowane poprzez ich modyfikacje potranslacyjne. Modyfikacje takie mogą obejmować kowalencyjne zmiany struktury cząsteczki, np. dodawanie dodatkowych grup funkcyjnych: fosforylacja, mirystylacja czy glikozylacja. Przykładowo związanie insuliny z odpowiednim receptorem uwalnia kaskadę fosforylacji wielu enzymów i w rezultacie syntezę glikogenu. Innym przykładem potranslacyjnych modyfikacji enzymów jest modyfikacja łańcucha polipeptydowego enzymu poprzez jego skracanie, przycinanie. Przykładowo chymotrypsyna, proteolityczny enzym trawienny, jest produkowany w trzustce i wydzielany do światła dwunastnicy w formie nieaktywnej, jako chymotrypsynogen. Tam następuje jego aktywacja poprzez skrócenie łańcucha polipeptydowego i uwolnienie aktywnej cząsteczki dojrzałego enzymu. Produkcja enzymu w formie proenzymu zabezpiecza trzustkę i inne tkanki, zanim dotrze do jelita, przed strawieniem. Nieaktywny prekursor enzymu nazywany jest proenzymem (dawniej zymogenem). Gdy aktywacja enzymu jest wieloetapowa, mówi się wtedy o preproenzymach. Przycinanie proenzymu do formy aktywnej zachodzi z udziałem innych enzymów lub czasami sam enzym jest w stanie katalizować reakcję aktywacji swoich form proenzymatycznych.
- Niektóre enzymy mogą stać się aktywne (nieaktywne), gdy znajdą się w innym środowisku, np. po przeniesieniu ze środowiska cytoplazmy o właściwościach redukujących do środowiska przestrzeni peryplazmatycznej o właściwościach utleniających, z wysokiego pH do niskiego pH itp. Przykładem może być hemaglutynina – glikoproteina o właściwościach antygenowych – na powierzchni wirusów grypy, która zmienia konformację, gdy znajdzie się w kwaśnym środowisku pęcherzyka komórkowego gospodarza, powodując aktywację wirusa.
Funkcje biologiczne
Enzymy są podstawą istnienia życia, jako że są niezbędne do zajścia niemalże każdej reakcji chemicznej z szybkością i wydajnością znacząco wysoką dla procesów biologicznych. Bez enzymów większość reakcji zachodziłaby zbyt wolno lub zbyt mało wydajnie, by miało to zauważalne w czasie znaczenie.
Większość enzymów, poprzez swój udział w reakcjach katabolicznych i anabolicznych, definiuje metabolizm komórki czy organizmu (elementami metabolizmu komórkowego są także enzymy zewnątrzkomórkowe, na przykład enzymy trawienne). Enzymy metaboliczne działają zwykle w grupach (kompleksach), w szeregu sekwencyjnie następujących po sobie reakcji, określanych mianem szlaków metabolicznych. Szlaki te często wykorzystują wspólne produkty pośrednie reakcji, przebiegają równolegle, prowadzą do tego samego produktu lub od tego samego substratu w skomplikowanej sieci zależności reakcji enzymatycznych. Dodatkowo regulacja tych szlaków, a także sygnalizacja wewnątrz- i zewnątrzkomórkowa także są zależne od enzymów.
Enzymy biorą udział także w reakcjach niemetabolicznych. Na przykład hydrolizująca ATP miozyna bierze udział w skurczach mięśni, a inne motory molekularne o aktywności enzymatycznej są odpowiedzialne za ruch na poziomie wewnątrzkomórkowym.
Enzymy są także ważnymi składnikami interakcji między organizmami czy komórkami. Leukocyty używają enzymów do zwalczania patogenów, z kolei bakterie patogenne używają ich podczas infekcji i do obrony przed elementami systemu immunologicznego. Także wirusy używają enzymów (kodowanych przez swój materiał genetyczny lub enzymów gospodarza) do swojej replikacji (np. odwrotna transkryptaza wirusa HIV), a także podczas infekcji i opuszczania komórek gospodarza (np. neuraminidaza wirusa grypy). Enzymy są także składnikami jadów i toksyn, używanych i wytwarzanych przez wiele organizmów.
Enzymy pełnią także bardziej wyszukane funkcje, wszędzie tam, gdzie potrzebna jest wydajna, specyficzna kataliza chemiczna. Na przykład bioluminescencja u świetlików wywołana jest przez enzym lucyferazę, a strzel bombardier, chrząszcz z rodzaju Brachynus w celach obronnych wykorzystuje peroksydazę do rozkładu nadtlenku wodoru, dzięki czemu może bronić się przed zagrożeniem, wyrzucając w jego stronę strumień wrzącej cieczy pod ciśnieniem.
Udział w chorobach i diagnostyce

Ponieważ aktywność enzymatyczna i jej ścisła kontrola są elementem homeostazy organizmu, defekt w jednym nawet enzymie (mutacja zaburzająca funkcję, nadprodukcja, za mała produkcja, delecja) czy mechanizmach kontroli jego aktywności, może prowadzić do stanu chorobowego lub śmierci organizmu.
Wrodzone uszkodzenia genów kodujących enzymy lub elementów kontroli ich aktywności mogą prowadzić do szeregu nieuleczalnych (albo leczonych wyłącznie objawowo) genetycznych chorób metabolicznych (to znaczy związanych z konkretnym szlakiem metabolicznym lub przemianami metabolicznymi). Należą do nich bloki metaboliczne (w tym choroby spichrzeniowe, złogowe), w których wskutek zakłócenia (braku) pracy enzymu, nie są metabolizowane konkretne substancje, co prowadzi do ich niedoboru lub gromadzenia się półproduktów, które często są toksyczne. Do takich chorób należą galaktozemia i homocystynuria. Ponieważ końcowe produkty danego szlaku są często inhibitorami enzymów jego wczesnych etapów, ich brak tym bardziej może kumulować szkodliwe półprodukty, produkowane przez nieregulowane enzymy (jak ma to miejsce w zespole Lescha-Nyhana). Także otyłość może być powodowana przez ogólnoustrojowe zaburzenie pracy enzymów metabolicznych.
Jedne enzymy często utrzymują aktywność innych na odpowiednim poziomie (np. poprzez rozkład ich nadmiaru). Gdy te pierwsze w jakiś sposób nie działają optymalnie, niekontrolowane już odpowiednio przez nie enzymy, mogą działać na szkodę własnych komórek organizmu. Może dojść do autolizy komórek i w następstwie do uszkodzenia tkanek. Przykładowo jedna z form rozedmy płuc jest powodowana niekontrolowaną aktywnością elastazy niszczącą strukturę tkanki.
Mutacje w niektórych enzymach mogą prowadzić do śmierci komórek lub powstawania nowotworów. Np. w enzymach zaangażowanych w naprawę materiału genetycznego lub kinaz tyrozynowych takich jak Bcr-Abl.
W chorobach wirusowych i bakteryjnych aktywność i inwazyjność patogenów często zależy od aktywności ich lub gospodarza enzymów. Także niektóre toksyny to enzymy ingerujące w metabolizm lub o aktywności litycznej, trawiennej, która może zaburzać funkcje życiowe zainfekowanego organizmu.
Enzymy w diagnostyce medycznej
Zmiany aktywności enzymów we krwi, często są odzwierciedleniem zmian patologicznych zachodzących w narządach. Nowoczesna diagnostyka enzymologiczna opiera się na założeniu, że uszkodzenie narządu pociąga za sobą uszkodzenie struktur komórkowych lub zmianę przepuszczalności błon komórkowych. Uszkodzenia błon powodują ucieczkę enzymów, zwiększając tym samym ich ilość w cieczach ustrojowych i wydalinach, takich jak: krew, płyn mózgowo-rdzeniowy, mocz, ciecze wysiękowe i przesiękowe, sok żołądkowy czy dwunastniczy. O lokalizacji, rozmiarach i intensywności uszkodzenia, i niewydolności narządu, można wnioskować nie tylko w przypadku wzrostu, ale również zmniejszeniu aktywności niektórych enzymów surowicy (tzw. sekrecyjnych). Poza stanami patologicznymi, na aktywność danego enzymu surowicy może wpływać intensywność ich produkcji przez organizm, a także zmiany ilościowe ich aktywatorów lub inhibitorów.
W latach 60. Richterich i Hess stworzyli kliniczny podział enzymów osocza na:
- sekrecyjne (wydzielnicze) – należą do nich m.in. czynniki krzepnięcia krwi oraz fibrynolizy, esterazy cholinowe, ceruloplazmina i lipaza lipoproteinowa. Po uszkodzeniu komórek wątroby następuje spadek ich aktywności, ponieważ ich ilość zależy od syntezy w rybosomach wątroby. W przypadku diagnostyki liczy się tylko dolna granica normy ich wydzielania;
- wskaźnikowe (indykatorowe) – pojawiają się w dużych ilościach po uszkodzeniu narządów (stąd ich potoczna nazwa – nekroenzymy). Zawartość enzymu wskaźnikowego jest zależna od jego ilości w uszkodzonej tkance, liczby uszkodzonych komórek i stopnia ich uszkodzenia, a także rozmieszczenia enzymów w różnych narządach. Do celów dydaktycznych można je podzielić na swoiste i nieswoiste narządowo. Diagnostycznie liczy się ich górna granica normy;
- ekskrecyjne (wydalnicze) – przeszkoda w normalnym odpływie różnych wydzielin ustrojowych, takich jak: żółć, sok trzustkowy, ciecz sterczowa czy ślina, powoduje zastój wydzieliny i przedostanie się enzymów do krwiobiegu. Do tej grupy należą enzymy soku trzustkowego (amylaza, lipaza, DNA-aza, RNA-aza, trypsyna, chymotrypsyna), żółci (fosfataza zasadowa, GGTP, leucyloaminopeptydaza – konstelacja żółci), fosfataza kwaśna płynu sterczowego, amylaza ślinianek i enzym gruczołów wydalniczych żołądka, czyli pepsyna.
Konwencja nazewnictwa
Zwykle nazwy enzymów pochodzą od substratów przetwarzanych w reakcji lub od samej reakcji przez nie obsługiwanych, do których dodaje się przyrostek -aza. Przykładowo celulaza to enzym rozkładający celulozę, a ligaza DNA to enzym ligujący cząsteczki DNA. Często nazwa jest dwuczłonowa, gdzie pierwszy człon określa reakcję, a drugi jej substrat, np. dehydrogenaza alkoholowa. Stąd różne enzymy obsługujące takie same reakcje lub przetwarzające te same substraty, nazywane izoenzymami, mimo tego, że chemicznie są cząsteczkami o różnym składzie aminokwasowym i właściwościach, będą miały takie same nazwy. Czasami nazwa enzymu nie pochodzi od reakcji obsługiwanej w warunkach fizjologicznych, in vivo, ale od jego aktywności in vitro. Przykładem jest izomeraza glukozowa, wykazująca in vitro aktywność izomeracji glukozy we fruktozę, ale in vivo będąca izomerazą ksylozową. Także zamiast od nazwy reakcji, enzymy są niekiedy nazywane od ogólnego procesu, jaki przeprowadzają, jak np. gyraza DNA (czyli topoizomeraza II), której nazwa pochodzi od greckiego słowa γύρος, czyli obracać i przyrostka -aza. Podczas przeprowadzanej reakcji enzym ten rzeczywiście musi obrócić nić DNA.
Z kolei niektóre cząsteczki biologiczne, mimo nazwy z przyrostkiem -aza, nie są enzymami lub definiowana przez nazwę aktywność nie jest aktywnością enzymatyczną. Przykładem są flipazy, których główna aktywność przenoszenia lipidów pomiędzy monowarstwami dwuwarstwy (proces flip-flop) nie jest aktywnością enzymatyczną (jest to proces fizyczny). Flipazy mogą mieć jednak aktywność enzymatyczną (np. ATP-azową) związaną lub nie z ich funkcją jako transporterów lipidów.
Duża część enzymów jest nazywana zwyczajowo, historycznie, jak np. pepsyna (nazwa pochodząca od greckiego słowa pepsis oznaczającego trawienie) czy papaina (enzym z owocu papai). Nazwy niektórych enzymów powstały także poprzez dodanie przyrostka -zym (tego samego co w słowie enzym) do ogólnej funkcji cząsteczki lub opisu, skrótu opisowego. Przykładami są lizozym (od lizujących właściwości wobec bakterii) czy granzym (ang. granule-associated enzyme, czyli enzym związany z ziarnistościami).
Enzymy restrykcyjne są nazywane według własnej nomenklatury. Ich nazwa składa się z litery nazwy rodzajowej i dwóch pierwszych liter nazwy gatunkowej mikroorganizmu-źródła enzymu, pisanymi kursywą, po których występują cyfry arabskie lub litery oznaczających szczep mikroorganizmu. Nazwa zakończona jest cyfrą rzymską wskazującą, jako który w kolejności chronologicznej został wyizolowany z danego organizmu dany enzym restrykcyjny. Na przykład EcoRI, to pierwszy wyizolowany enzym restrykcyjny z bakterii Escherichia coli, szczep RY13.
Część enzymów jest natomiast bardziej znana pod skrótami pochodzącymi od ich pełnych nazw, jak RuBisCO (czyli karboksylaza oksygenaza rybulozo-1,5-bisfosforanu, w ang. ribulose-1,5-bisphosphate carboxylase/oxygenase), albo od skrótu nazwy opisowej, jak BACE2 (z ang. beta-site APP-cleaving enzyme 2, czyli enzym tnący prekursorowe białko amyloidu 2).
W celu uregulowania i ujednoznacznienia nazewnictwa enzymów, Komitet Nazewnictwa (ang. Nomenclature Committee) Międzynarodowej Unii Biochemii i Biologii Molekularnej (ang. International Union of Biochemistry and Molecular Biology) w latach 1956–1972 opracował dla nich nomenklaturę naukową – numer EC. Według niej, każdy enzym jest opisany przez ciąg czterech segmentów cyfr, oddzielonych od siebie kropką, poprzedzonych literami „EC” (Enzyme Commission lub Enzyme Catalogue): EC x.xx.xx.xx.
Pierwsza cyfra dzieli enzymy, według mechanizmu reakcji przez nie katalizowanych, na siedem głównych klas:
- EC 1 oksydoreduktazy: katalizują reakcje utleniania i redukcji,
- EC 2 transferazy: przenoszą grupy funkcyjne,
- EC 3 hydrolazy: katalizują hydrolizę różnych wiązań,
- EC 4 liazy: rozcinają różne wiązania na drodze innej niż hydroliza czy utlenianie,
- EC 5 izomerazy: katalizują zmiany izomeracyjne cząsteczek,
- EC 6 ligazy: łączą cząsteczki wiązaniami kowalencyjnymi,
- EC 7 translokazy: katalizują ruch jonów i cząsteczek przez błony lub ich rozdział wewnątrz błon.
Następne liczby numeru EC oddzielone kropkami klasyfikują dany enzym odpowiednio do podklasy i podpodklasy, natomiast ostatnia liczba określa miejsce enzymu w podpodklasie.
W nazewnictwie enzymów zaleca się stosowanie numeru EC, jednak dopuszcza się stosowanie nazw zwyczajowych z przyrostkiem -aza, gdzie pierwszy człon określa reakcję, a drugi jej substrat, a także innych ogólnie przyjętych nazw roboczych i zwyczajowych, co także jest regulowane stosownymi zaleceniami.
Zastosowanie przemysłowe
Enzymy są stosowane w przemyśle chemicznym, spożywczym i innych, głównie jako niezwykle specyficzne, bezpieczne w użyciu katalizatory. Jakkolwiek ich wadą jest wrażliwość na skrajne warunki (np. temperatura, pH), niestabilność w środowiskach innych niż wodne (np. rozpuszczalników organicznych) oraz stopniowa degradacja podczas użytkowania. Także wysoka specyficzność, istotna z punktu biologicznego, w przemyśle jest ograniczeniem ich uniwersalności. Stąd inżynieria białka jest dynamicznie rozwijającą się dziedziną nauki, zajmującą się badaniem i projektowaniem enzymów o nowych właściwościach lub poprawionej wydajności czy stabilności. Aktualne podejście do tego zagadnienia to ukierunkowane projektowanie lub ewolucja in vitro. Obecnie enzymy produkowane są na skalę przemysłową, głównie z zastosowaniem mikroorganizmów modyfikowanych genetycznie.
| Zastosowanie | Używane enzymy | Cel | ||
|---|---|---|---|---|
Przemysł piekarniczy 
Łańcuchy skrobi są rozkładane na krótsze cukry przez alfa-amylazę
|
Grzybowe alfa-amylazy | Katalityczne przyspieszenie rozkładu skrobi z mąki na krótsze cukry. Te z kolei są wykorzystywane przez drożdże piekarnicze zawarte w cieście, wskutek czego produkowany jest dwutlenek węgla, spulchniający ciasto. Używane w produkcji pieczywa białego, bułek itp. | ||
| Proteazy | Producenci ciastek używają ich do obniżenia poziomu białka w mące. | |||
| Jedzenie dla niemowląt | Trypsyna | Do wstępnego nadtrawiania jedzenia. | ||
| Browarnictwo, gorzelnictwo i przemysł winiarski | Enzymy jęczmienne uwalniane są podczas przyrządzania zacieru przy produkcji piwa. | Rozkładają one skrobię i białka, uwalniając cukry proste i aminokwasy, służące następnie drożdżom w fermentacji. | ||
| Przemysłowo produkowane enzymy jęczmienne i drożdżowe | Używane jako substytuty naturalnych | |||
| Amylazy, glukanazy, proteazy | Rozkładają polisacharydy i białka zawarte w słodzie czy zacierze. | |||
| Betaglukozydaza | Poprawia proces filtrowania. | |||
| Amyloglukozydaza | Produkcja trunków niskokalorycznych. | |||
| Proteazy | Usuwają zmętnienia produktu. | |||
| Dekarboksylaza kwasu α-acetomlekowego (ALDC) | Poprawia wydajność fermentacji usuwając nadmiar biacetylu | |||
| Soki owocowe | Celulazy, pektynazy | Klarują soki (rozkład celulozy i pektyn). | ||
| Mleczarstwo | Podpuszczka, izolowana z żołądków młodych przeżuwaczy (cielęta, owce). | Produkcja serów, używane do hydrolizy białek. | ||
| Enzymy pochodzenia mikrobiologicznego | Zastępują te pochodzenia zwierzęcego. | |||
| Lipazy | Stosowane w produkcji sera Roquefort by usprawnić ich dojrzewanie. | |||
| Laktazy | Rozkład laktozy do glukozy i galaktozy. | |||
| Zmiękczacze mięsa | Papaina | Rozmiękczanie mięsa przy gotowaniu. | ||
|
Przemysł skrobiowy
|
Amylazy, amyloglukozydazy i glukoamylazy | Rozkład skrobi do glukozy i produkcja cukrów inwertowanych. | ||
| Izomeraza glukozowa | Konwersja glukozy we fruktozę przy produkcji wysokofruktozowych syropów ze skrobi. Syropy te są używane do słodzenia i są słodsze niż sacharoza, a mniej kaloryczne. | |||
Przemysł papierniczy
Wytwórnia papieru w Południowej Karolinie
|
Amylaza, ksylanaza, celulaza i ligninaza | Amylazy rozkładają skrobię w celu zmniejszenia lepkości, wspomagają regulację gramatury papieru i jego pokrycia. Ksylanazy redukują potrzebę wybielania. Celulazy wygładzają włókna, wspomagają schnięcie oraz usuwanie pigmentów (przy przeróbce makulatury). Ligninazy degradują ligniny i wspomagają zmiękczanie papieru. | ||
| Przemysł biopaliwowy | Celulaza | Używana do rozkładu celulozy do cukrów, które mogą być wykorzystane w procesie fermentacji przy produkcji etanolu celulozowego. | ||
| Ligninaza | Używana do rozkładu ligniny | |||
| Biodetergenty | Proteazy, pierwotnie pozyskiwane z wydzielających je na zewnątrz komórek bakterii. | Używane w praniu wstępnym i namaczaniu, a także podczas prania zasadniczego, gdzie wspomagają usuwanie plam pochodzenia białkowego z ubrań. | ||
| Amylaza | Do usuwania plam ze skrobi. | |||
| Lipaza | Wspomagają usuwanie plam z tłuszczu. | |||
| Celulaza | Używana w biologicznych zmiękczaczach do tkanin. | |||
| Płyny do mycia soczewek kontaktowych | Proteazy | Do odbiałczania szkieł i zapobiegania zakażeniom. | ||
| Przemysł gumowy | Katalaza | Do wytwarzania tlenu z nadtlenków, by przetwarzać lateks w gumy spienione. | ||
| Przemysł fotograficzny | Proteazy | Rozkład żelatyny pokrywających filmy fotograficzne przy odzyskiwaniu z nich srebra. | ||
Biologia molekularna, biochemia i pokrewne, a także analityka laboratoryjna i kliniczna 
Odcinek DNA w formie podwójnej helisy
|
M.in. enzymy restrykcyjne, ligazy DNA, polimerazy DNA i RNA, proteazy, DNazy, RNazy, enzymy szlaków metabolicznych i inne. | Używane m.in.: w manipulacjach materiałem genetycznym, badaniach metabolizmu, proteomice, w diagnozowaniu klinicznym i kryminalnym, jako markery i indykatory, oraz wiele innych. |



Uwagi
Linki zewnętrzne
- Enzyme Structures Database – baza danych trójwymiarowych struktur enzymów w Protein Data Bank (ang.);
- ExPASy enzyme – baza danych, linki do baz sekwencji Swiss-Prot, wejścia do innych baz danych (ang.);
- KEGG: Kyoto Encyclopedia of Genes and Genomes – graficzna i tekstowa dokumentacja szlaków metabolicznych oraz enzymów (ang.);
- MetaCyc – baza danych enzymów i szlaków metabolicznych (ang.);
- BRENDA – baza danych kompletnych informacji o enzymach; płatna dla użytkowników komercyjnych (ang.);
- Enzyme spotlight – miesięcznik przygotowywany przy udziale Europejskiego Instytutu Bioinformatyki na temat wybranych białek (ang.).