

Historia odkrycia stwardnienia guzowatego i badań nad tą chorobą liczy dopiero niecałe 200 lat. Stwardnienie guzowate (tuberous sclerosis, tuberous sclerosis complex, TSC) jest rzadką, wielonarządową chorobą genetyczną, w której rozwijają się łagodne guzy mózgu i guzy innych ważnych życiowo narządów: nerek, serca, oczu, płuc i skóry. Zespół objawów może obejmować napady drgawkowe, opóźnienie rozwoju, zaburzenia behawioralne i schorzenia dermatologiczne, a także objawy wynikające z zajęcia płuc i nerek. TSC może być spowodowane mutacją w jednym z dwóch genów: TSC1 i TSC2, kodujących, odpowiednio, hamartynę i tuberynę. Oba geny należą do genów supresorowych (antyonkogenów), gdyż funkcją kodowanych przez nie białek jest regulacja cyklu komórkowego i procesu różnicowania komórek. W przeszłości zachorowania na tę chorobę traktowano jak ciekawe przypadki kazuistyczne; obecnie, badaniom nad patogenezą TSC przypisuje się istotne znaczenie w poznawaniu procesu nowotworzenia i supresji nowotworów.
Historię badań nad stwardnieniem guzowatym można podzielić na cztery okresy. Pod koniec XIX wieku, wybitni lekarze pracujący w największych szpitalach klinicznych pierwsi opisali korowe i dermatologiczne objawy choroby; uczonych tych uhonorowano eponimicznymi określeniami – "choroby Bourneville'a" i "znamienia Pringle'a". Na początku XX wieku skojarzono te objawy jako charakterystyczne dla jednej jednostki chorobowej. Stwierdzono, że choroba może zajmować inne narządy i zdano sobie sprawę ze zmienności obrazu klinicznego i ciężkości choroby. Pod koniec XX wieku dokonał się wielki postęp w technikach obrazowania układu nerwowego, odkryto też dwa geny, których mutacje wywołują chorobę. W końcu, na początku XXI wieku miały miejsce pierwsze odkrycia pozwalające zrozumieć patomechanizm choroby na poziomie subkomórkowym, otwierające równocześnie nowe możliwości niechirurgicznego leczenia.
XIX wiek
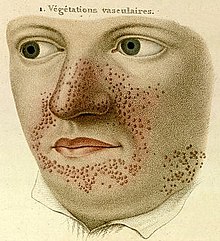
- 1835
- Francuski dermatolog Pierre François Olive Rayer opublikował atlas chorób skóry. Zawiera on 22 duże, kolorowe plansze z 400 rycinami przedstawionymi w usystematyzowany w sposób. Na stronie 20. rycina 1 przedstawia rysunek, który jest uważany za najwcześniejszy opis stwardnienia guzowatego. Przy rysunku opisanym "végétations vasculaires" Rayer dodał komentarz – "są to małe naczynia krwionośne grudkowatego wyglądu, szeroko rozprzestrzenione na nosie i wokół ust". W atlasie nie wspomniano nic o objawach towarzyszących przedstawionym zmianom skórnym.
- 1850
- Angielscy dermatolodzy Thomas Addison i William Gull opisali na łamach "Guy's Hospital Reports" przypadek 4-letniej dziewczynki z "niezwykłą wysypką obejmującą w poprzek nos i część obu policzków", który określili jako "vitiligoidea tuberosa" (bielaczość guzowata).
- 1862
- Niemiecki lekarz Friedrich Daniel von Recklinghausen, pracujący wówczas jako asystent Rudolfa Virchowa w Instytucie Anatomii Patologicznej w Berlinie zaprezentował Berlińskiemu Towarzystwu Położniczemu przypadek serca noworodka z kilkoma guzami, który "zmarł zaledwie po kilku [samodzielnych] oddechach". Nazwał te guzy "myomata". Jeden z nich był "wielkości gołębiego jaja". Zauważył także, że mózg miał "dużą liczbę stwardnień". Były to prawie z całkowitą pewnością mięśniaki prążkowanokomórkowe serca oraz guzy korowe występujące w stwardnieniu guzowatym. Recklinghausen nie zdołał rozpoznać odmiennej jednostki chorobowej, odnosząc się do tego przypadku jak do anatomopatologicznej ciekawostki. Nazwisko Recklinghausena zostało później powiązane z nerwiakowłókniakowatością po klasycznej publikacji z 1881.
- 1864
- Niemiecki patolog Rudolf Virchow opublikował trzytomową pracę o nowotworach, opisującą przypadek dziecka chorego na stwardnienie guzowate ze zmianami narządowymi w mózgu i sercu. Praca Virchowa zawiera pierwszą udokumentowaną wskazówkę dziedziczności choroby: siostra dziecka zmarła z powodu guza mózgu.
- 1880
- Francuski neurolog Désiré-Magloire Bourneville miał okazję spotkać się z chorobą, której eponim zawiera jego nazwisko. Bourneville pracował jako nieoficjalny asystent Jeana Martina Charcota w La Salpêtrière. W czasie zastępstwa swojego nauczyciela Lois J.F. Delasiauve, odwiedzał Marie, 15-letnią dziewczynkę z opóźnieniem psychoruchowym, padaczką oraz zlewnymi naczyniowo-grudkowymi wypryskami na policzkach i czole (rozpoznanymi błędnie jako trądzik różowaty). W wywiadzie miała historię napadów padaczkowych od dzieciństwa, a w wieku 3 lat trafiła do szpitala dziecięcego i tam określono jej przypadek jako beznadziejny. Miała trudności z uczeniem się i nie potrafiła ani chodzić, ani mówić. W czasie opieki Boourneville'a Marie miała coraz częstsze napady padaczkowe, które następowały jeden po drugim. Leczono ją alkoholowymi napojami z chininą, bromkiem kamfory, azotynem amylu oraz pijawkami przykładanymi za uszami. 7 maja 1879 Marie zmarła w szpitalu. Sekcja zwłok wykazała obecność twardych, zwartych guzów w zakrętach mózgu, które Bourneville nazwał sclérose tubéreuse des circonvolutions cérébrales (stwardnieniem guzowatym zakrętów mózgu). Nazwa choroby wymyślona przez Bourneville'a pochodziła od greckich słów tuber, oznaczającego bulwę lub ziemniak, i skleros, znaczącego „twardy”. Bourneville wywnioskował, że zmiany w mózgu są źródłem napadów padaczkowych. W dodatku w obu nerkach znaleziono twarde białe masy wielkości orzecha włoskiego.
- 1881
- Niemiecki lekarz Hartdegen opisał przypadek dwudniowego noworodka zmarłego w stanie padaczkowym. Badanie pośmiertne ujawniło małe guzy w komorach bocznych mózgu oraz obszary stwardnienia korowego, które Hartdegen nazwał glioma gangliocellulare cerebri congenitum.

- 1881
- Bourneville i Édouard Brissaud przebadali czteroletniego chłopca w szpitalu La Bicétre. Podobnie jak poprzednio, pacjent miał guzy korowe, padaczkę i trudności w nauce. Dodatkowo stwierdzili u niego szmer serca, a w badaniu pośmiertnym okazało się, że miał małe twarde guzki w ścianach komór oraz w mózgu (guzki subependymalne), a także małe guzy w nerkach (przypuszczalnie naczyniakomięśniakotłuszczaki). Do 1889 roku Bourneville opisał jeszcze dziesięcioro pacjentów ze stwardnieniem guzowatym.
- 1885
- Francuscy lekarze Félix Balzer i Pierre Eugène Ménétrier opisali przypadek "adénomes sébacés de la face et du cuir" (gruczolaki łojowe twarzy i głowy). Obecnie wiadomo, że to określenie nie było poprawne, ponieważ nie są to ani gruczolaki, ani nie wywodzą się z gruczołów łojowych. Wysypka grudkowa jest obecnie znana jako naczyniakowłókniak twarzy.
- 1885
- Francuscy dermatolodzy François Henri Hallopeau i Émile Leredde opisali przypadek zmian adenoma sebaceum o twardej i włóknistej konsystencji. Pierwsi opisali plamy szagrynowe, a później stwierdzili związek wysypki na twarzy i padaczki.
- 1890
- Szkocki dermatolog pracujący w Londynie, John James Pringle, opisał przypadek 25-letniej kobiety z obniżoną inteligencją, szorstkimi zmianami na rękach i nogach, i grudkowatą wysypką na skórze twarzy określoną później od jego nazwiska guzkami Pringle'a. Pringle w swojej pracy przywołał również pięć wcześniejszych doniesień, z których dwa były niepublikowane. Gruczolak łojowy (adenoma sebaceum) Pringle'a stał się powszechnie stosowanym eponimem na określenie tego typu zmian skórnych twarzy.
Początek XX wieku


- 1901
- Włoski lekarz Giovanni Battista Pellizzi zbadał patologiczne zmiany w mózgowiu chorych. Stwierdził ich dysplastyczny charakter, obecność heterotopii korowych i obszarów nieprawidłowej mielinizacji. Pellizzi sklasyfikował guzy do dwóch typów: typ 1 (o gładkiej powierzchni) i typ 2 (ze środkowym zagłębieniem).
- 1903
- Niemiecki lekarz Richard Kothe opisał włókniaki okołopaznokciowe, opisane ponownie przez holenderskiego lekarza Joannesa Koenena w 1932 (od którego wzięły nazwę guzków Koenena).
- 1906
- Austriacki neurolog Alfred Walter Campbell pracujący w Anglii, uznał zmiany występujące w mózgu, skórze, sercu i nerkach za części obrazu klinicznego jednej choroby. Także jako pierwszy opisał patologiczne zmiany w oku w przebiegu TSC. Na podstawie swojego przeglądu historii 20 pacjentów jako pierwszy zaproponował diagnostyczną triadę objawów, powszechniej przypisywaną Vogtowi.
- 1907
- Franciszek Krzyształowicz przedstawił pierwszy w polskim piśmiennictwie opis choroby.
- 1908
- Niemiecki neurolog dziecięcy Heinrich Vogt ustalił kryteria diagnostyczne stwardnienia guzowatego, bezspornie potwierdzając związek zmian skórnych na twarzy z neurologicznymi następstwami guzów korowych. Triada Vogta: epilepsja-idiotyzm-adenoma sebaceum obowiązywała przez 60 lat, zanim badania Manuela Gómeza nie dowiodły, że mniej niż 1/3 pacjentów z TSC prezentuje wszystkie trzy objawy.
- 1910
- Joseph Kirpicznik jako pierwszy stwierdził genetyczny charakter choroby. Opisał przypadek jedno- i dwujajowych bliźniąt, a także jednej rodziny, w której przypadki choroby występowały w trzech pokoleniach.
- 1911
- Edward Sherlock, adwokat i wykładowca biologii doniósł w swojej książce o dziewięciu przypadkach osób "ograniczonych umysłowo". Ukuł termin epiloia, zbitkę greckich słów epilepsia oraz anoia (bezmyślny). To określenie nie jest już używane jako synonim TSC. Genetyk Robert James Gorlin zasugerował w 1981, że mógłby to być użyteczny akronim od epilepsja, low intelligence (niska inteligencja) oraz adenoma sebaceum (gruczolak łojowy).
- 1912
- Polscy neurolodzy Kazimierz Orzechowski i W. Nowicki opublikowali pracę, w której podkreślono podobieństwo chorób Recklinghausena i Bourneville'a.
- 1913
- H. Berg jako jeden z pierwszych uznał TSC za chorobę dziedziczną, opisując zachorowania w dwóch i trzech generacjach rodzin.
- 1914
- P. Schuster opisał pacjenta z adenoma sebaceum i epilepsją, ale o normalnej inteligencji. Taki niepełny fenotyp choroby określa się jako postać poronną TSC albo forme fruste. Schuster opisał ponownie ogniska skóry szagrynowej (wcześniejsze odkrycie François Henriego Hallopeau i Émile'a Lereddego nie było mu znane).
- 1918
- Francuski lekarz René Lutembacher opisał pierwszy przypadek torbielowatych zmian w płucach w przebiegu TSC. 36-letnia pacjentka zmarła z powodu obustronnej odmy opłucnowej. Lutembacher stwierdził, że torbiele i guzki w tkance płucnej były przerzutami włókniakomięsaka nerki. Obecnie to powikłanie TSC, spotykane znacznie częściej u kobiet, określa się jako limfangioleiomiomatozę (LAM).
- 1920
- Duński okulista Jan van der Hoeve opisał zmiany o charakterze hamartomata siatkówki (phakoma). Powiązał TSC i nerwiakowłókniakowatość w jedną grupę chorób, które określił jako fakomatozy (później używano też terminu zespołów nerwowoskórnych).
- 1924
- H. Marcus stwierdził, że charakterystyczne dla TSC zmiany, takie jak wewnątrzczaszkowe zwapnienia, są widoczne na radiogramie.
Lata 1925-1975
- 1932
- MacDonald Critchley i Charles J.C. Earl przebadali 29 pacjentów ze stwardnieniem guzowatym w ośrodkach psychiatrycznych. Zaobserwowali charakterystyczne zachowania: ruchy rąk, dziwne postawy ciała i powtarzanie wykonywanych czynności (stereotypie ruchowe) – obecnie określane jako autystyczne. Było to 11 lat, zanim Leo Kanner wprowadził pojęcie autyzmu. Odnotowali również obecność białych, hipomelanotycznych plam na skórze tych pacjentów.
- 1933
- Edward Alfred Cockayne w monografii swojego autorstwa zwrócił uwagę na dominujący sposób dziedziczenia TSC.
- 1934
- N.J. Berkwitz i L.G. Rigler wykazali, że możliwe jest rozpoznanie stwardnienia guzowatego przy pomocy pneumoencefalografii, w której można uwidocznić niezwapniałe guzki podkorowe. Obraz na ścianach komór bocznych mózgu przypominał "krople wosku ściekające z palącej się świecy".
- 1935
- Gunther i Penrose opublikowali pracę podsumowującą ich obserwacje nad dziedziczeniem TSC.
- 1942
- Sylvan E. Moolten zaproponował termin "zespół stwardnienia guzowatego" (tuberous sclerosis complex – TSC), który jest obecnie preferowany w piśmiennictwie anglojęzycznym. Nazwa odzwierciedla wielonarządową naturę choroby. Moolten wprowadził też szereg pojęć określających charakter zmian w stwardnieniu guzowatym: "podstawową zmianą jest hamartia, przekształcająca się w zmianę guzowatą (hamartoma) lub prawdziwy nowotwór (hamartoblastoma)".
- 1954
- Norweski patolog Reidar Eker wyhodował szczep szczurów Wistar z predyspozycją do rozwoju gruczolaków nadnerczy. Szczur Ekera został cennym modelem raka dziedziczonego w sposób dominujący.
- 1966
- Phanor Perot i Bruce Weir zostali pionierami w chirurgicznej interwencji w przypadku padaczki występującej w stwardnieniu guzowatym. Spośród siedmiu pacjentów poddanych korowej resekcji guzów, dwóch zostało uwolnionych od napadów. Wcześniej tylko czterech pacjentów zostało poddanych leczeniu chirurgicznemu w padaczce związanej ze stwardnieniem guzowatym.
- 1967
- J.C. Lagos i Manuel Rodríguez Gómez przebadali 71 przypadków stwardnienia guzowatego i wykazali, że w 38% osoby z tą chorobą mają prawidłowy iloraz inteligencji.
- 1971
- Amerykański genetyk Alfred Knudson postawił hipotezę "dwóch uderzeń" wyjaśniającą powstawanie retinoblastoma zarówno u dzieci, jak i dorosłych. Według tej teorii nowotwór powstawał u dzieci z wrodzonymi mutacjami linii germinalnych, do których dołączyły się mutacje somatyczne we wczesnym okresie życia. Ten model odnosi się do wielu innych przypadków, w tym związanych z genami supresorowymi, takimi jak TSC. W latach 80. badania Knudsona nad szczurami Ekera umocniły tę hipotezę.
- 1975
- Giuseppe Pampiglione i E. Pugh, w artykule w The Lancet, donosili, że do 69% pacjentów ze stwardnieniem guzowatym zgłaszanych jest z drgawkami noworodkowymi.
- 1975
- Riemann jako pierwszy zastosował USG do badania zajętych w przebiegu stwardnienia guzowatego nerek 35-letniej kobiety z przewlekłą niewydolnością nerek.
Ostatnia kwarta XX wieku

- 1976
- Tomografia komputerowa (CT, wynaleziona w 1972) głowy okazała się doskonałym narzędziem do diagnozowania nowotworów mózgu u dzieci, także w przypadku tych występujących w stwardnieniu guzowatym.
- 1979
- Manuel Gómez opublikował monografię: Tuberous Sclerosis, która pozostaje standardowym podręcznikiem przez 3 edycje w ciągu 20 lat. Książka jako pierwsza opisywała pełne spektrum kliniczne stwardnienia guzowatego i ustanawiała nowy zestaw kryteriów diagnostycznych, które zastąpiły triadę Vogta.
- 1982
- Kenneth Arndt leczył z powodzeniem naczyniakowłókniaka twarzy za pomocą lasera argonowego.
- 1983
- Pozytonowa emisyjna tomografia komputerowa (PET, wynaleziona w 1981) została porównana z elektroencefalografią (EEG) i CT. Okazało się, że jest w stanie zlokalizować korowe guzy odpowiadające za napady padaczkowe, który normalnie nie zostałyby wykryte.
- 1984
- Stwierdzono, że napad drgawek u niemowlęcia z TSC poprzedziło ogniskowe wyładowanie w EEG.
- 1985
- Obrazowanie rezonansu magnetycznego (MRI, odkryte w 1980) zostało po raz pierwszy użyte w stwardnieniu guzowatym do zidentyfikowania zmienionych obszarów mózgu chorej dziewczynki.
- 1987
- Czułość i swoistość MR zostały ocenione na wyższe niż w przypadku CT. W badaniu, które objęło 15 pacjentów, MR pozwoliło zidentyfikować guzki podwyściółkowe wpuklające się do komór bocznych u 12 pacjentów, zaburzenie prawidłowej architektoniki korowej u 10 pacjentów (w związku z występowaniem guzów korowych), poszerzonych komór u 5 pacjentów i pozwoliło na rozróżnienie astrocytoma od łagodnych guzków subependymalnych u jednego pacjenta.
- 1987
- Stwierdzono, że obrazowanie MR pozwala na prognozowanie ciężkości klinicznego przebiegu choroby (padaczki i opóźnienia rozwoju). Badanie na 25 pacjentach wykazało istnienie korelacji z liczbą guzów korowych. W przeciwieństwie do MR, CT nie wykazało takiej przydatności, ale było skuteczniejsze w wykrywaniu zwapniałych zmian w mózgu.
- 1987
- Analiza sprzężeń przeprowadzona na 19 rodzinach z TSC pozwoliła ustalić przypuszczalny locus genu odpowiedzialnego za TSC na chromosomie 9, w pobliżu locus dla białek determinujących grupy krwi AB0. Ponieważ wielu chorych nie wykazywało zmian w tym locus, kontynuowano badania nad poszukiwaniem dodatkowych genów zaangażowanych w patogenezę TSC.
- 1988
- Guzy korowe wykryte w MR okazały się dokładnie odpowiadać utrwalonym miejscom ognisk w EEG w badaniu na szóstce dzieci z TSC. W szczególności guzy kory czołowej były związane z drgawkami o mniejszej podatności na leczenie.
- 1990
- Stwierdzono, że wigabatryna jest wysoce skutecznym lekiem w leczeniu drgawek u dzieci, zwłaszcza u dzieci z TSC. Po odkryciu w 1997 roku, że możliwym działaniem niepożądanym leku jest ciężkie trwałe ograniczenie pola widzenia, zastosowanie wigabatryny w monoterapii ograniczono do tej grupy pacjentów.
- 1992
- Analiza sprzężeń pozwoliła ustalić locus drugiego genu związanego z TSC na 16p13.3, w pobliżu genu, którego mutacje odpowiadają za wielotorbielowatość nerek typu 1 (PKD1).
- 1993
- Zespół naukowców European Chromosome 16 Tuberous Sclerosis Consortium korzystając z prac naukowców, którzy wcześniej zmapowali region 16p13.3 w poszukiwaniu genu APKD1 sklonował gen TSC2 kodujący tuberynę, i ogłosił wyniki na łamach Cell.
- 1994
- Odkryto, że szczury Ekera są zwierzęcym modelem stwardnienia guzowatego; są nosicielami mutacji w szczurzym odpowiedniku genu TSC2.
- 1995
- Okazało się, że MRI w sekwencji FLAIR (ang. fluid attenuated inversion recovery) jest metodą znacznie lepszą w wykrywaniu małych guzów, zwłaszcza podkorowych w porównaniu do standardowego obrazowania w sekwencji T2.
- 1997
- Zespół naukowców TSC1 Consortium ogłosił w Science sklonowanie genu TSC1 kodującego hamartynę.
- 1997
- Odkryto, że gen PKD1, który odpowiada za przekazywany autosomalnie dominująco zespół wielotorbielowatych nerek oraz gen TSC2 sąsiadują ze sobą na chromosomie 16p13.3. Zespół utworzony w Institute of Medical Genetics in Wales przebadał 27 niepowiązanych pacjentów ze stwardnieniem guzowatym oraz z wielotorbielowatością nerek. Wywnioskowano, że poważne przypadki chorób nerek u osób ze stwardnieniem guzowatym są spowodowane przez sąsiedztwo delecji w genach TSC2 i PKD1. Zauważono również, że choroba była odmienna od wielotorbielowatości nerek (zaczynała się wcześniej i miała cięższy przebieg), a także TSC1 bez znaczącej choroby torbielowatej.
- 1997
- Patrick Bolton i Paul Griffiths przebadali 18 pacjentów z TSC, z których połowa miała jakąś postać autyzmu. Znaleźli oni silną korelację pomiędzy obecnością guzów w płacie skroniowym a wystąpieniem autyzmu.
- 1998
- Tuberous Sclerosis Consensus Conference (Konferencja Konsensusowa dotycząca Stwardnienia Guzowatego) wydała poprawione kryteria diagnostyczne, które są obecnym standardem.
- 1998
- Włoski zespół użył magnetoencefalografii do przebadania trzech pacjentów ze stwardnieniem guzowatym i częściową(?) epilepsją. W połączeniu z MRI zdołali oni znaleźć powiązanie pomiędzy guzowatymi obszarami w mózgu, nieprawidłową funkcją neuronalną, a źródłami napadów padaczkowych. Późniejsze badania potwierdziły, że MEG jest lepszą metodą niż EEG w identyfikowaniu guzów odpowiedzialnych za padaczkę, które mogą stać się kandydatami do resekcji (wycięcia)
XXI wiek

- 2001
- Wieloośrodkowe badania kohortowe na 224 pacjentach zostały przeprowadzone dla sprawdzenia mutacji oraz ciężkości przebiegu choroby. Osoby z TSC1 mają lżejszą postać choroby niż inni z TSC2. Zdarza im się mniej napadów padaczkowych oraz są mniej upośledzeni umysłowo. Niektóre objawy TSC były rzadkie lub nawet nieobecne u osób z TSC1. Wnioskuje się, że "zarówno mutacje germinalne, jak i somatyczne wydają się mniej powszechne w TSC1 niż w TSC2".
- 2002
- Kilka grup naukowców zbadało, w jaki sposób produkty genów TSC1 i TSC2 (tuberyna i hamartyna) łącznie działają w blokowaniu szlaku sygnalizacyjnego kinazy mTOR (kinazy-ssaczego celu rapamycyny, mammalian target of rapamycin). Ten istotny szlak sygnalizacyjny reguluje proliferację komórek i proces supresji nowotworzenia.
- 2002
- Udowodniono, że terapia rapamycyną (sirolimusem) prowadziła do zmniejszenia objętości guzów u szczurów Ekera (zwierzęcy model mutacji TSC2 człowieka) oraz mysich (TSC1) modeli stwardnienia guzowatego.
- 2006
- Opublikowano obiecujące rezultaty małych badań klinicznych, w których terapia rapamycyną powodowała zmniejszenie naczyniakomięśniakotłuszczaków i gwiaździaków. Rozpoczęto większe, wieloośrodkowe badania nad leczeniem limfangioleiomiomatozy i naczyniakomięśniakotłuszczaków (AML) nerek za pomocą rapamycyny, glejaków olbrzymiokomórkowych za pomocą ewerolimusu, pochodnej rapamycyny.
Bibliografia
- Acierno, Louis J: The History of Cardiology. Taylor & Francis, 1994, s. 427. ISBN 1-85070-339-6.
- Historical Backgound. W: Curatolo, Paolo (red.): Tuberous Sclerosis Complex : From Basic Science to Clinical Phenotypes. MacKeith Press, 2003, s. 1-10. ISBN 1-898683-39-5.
- Gómez MR. History of the tuberous sclerosis complex. „Brain & Development”. 17. suppl, s. 55-7, 1995.
- Rott HD, Mayer K, Walther B, Wienecke R: Zur Geschichte der Tuberösen Sklerose (The History of Tuberous Sclerosis). Tuberöse Sklerose Deutschland e.V, 2005. [dostęp 2007-09-01]. (niem.).