
| epidermolysis bullosa | |

| |
| Klasyfikacje | |
| ICD-10 | |
|---|---|
| Q81.0 |
Pęcherzowe oddzielanie naskórka proste |
| Q81.1 |
Pęcherzowe oddzielanie naskórka, postać śmiertelna |
| Q81.2 |
Pęcherzowe oddzielanie naskórka, postać dystroficzna |
| Q81.8 |
Inne oddzielanie pęcherzowe naskórka |
| Q81.9 |
Pęcherzowe oddzielanie naskórka, nie określone |
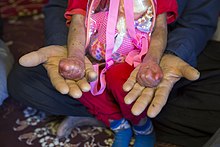
Pęcherzowe oddzielanie się naskórka (łac. epidermolysis bullosa) – grupa pęcherzowych chorób skóry uwarunkowanych genetycznie (genodermatoz). Wspólną cechą tych chorób jest występowanie pęcherzy wskutek urazów mechanicznych.
Epidemiologia
Choroby grupy epidermolysis bullosa są niezwykle rzadkie. Szacuje się, że występują z częstością 2/100 000 osób.
Etiopatogeneza i klasyfikacja
Wyróżnia się trzy główne grupy, w zależności od miejsca tworzenia się pęcherza:
- w obrębie naskórka (EB simplex)
- w obrębie błony podstawnej (EB junctionalis)
- poniżej błony podstawnej, w skórze właściwej (EB epidermolytica).
Postęp w biologii molekularnej umożliwił niedawno dermatologom na ujednolicenie klasyfikacji EB, co doprowadziło do konsensusu w kwestii nazewnictwa i podziału.
| Grupa | Typy | Uszkodzony gen | Nieprawidłowe białko | OMIM |
|---|---|---|---|---|
| EB simplex (EBS) |
EBS Weber-Cockayne EBS Koebner EBS Dowling-Meara EBS z dystrofią mięśniową |
KRT 5 i KRT 14 KRT 5 i KRT 14 KRT 5 i KRT 14 PLEC 1 |
Keratyna 5 i 14 Keratyna 5 i 14 Keratyna 5 i 14 Plektyna |
OMIM 131800 OMIM 131900 OMIM 131760 OMIM 226670 |
| EB junctionalis (EBJ) |
EBJ Herlitz EBJ non-Herlitz EBJ z atrezją odźwiernika |
LAMB3, LAMC2, LAMA3 COL17A1, LAMB3, LAMC2, LAMA3 ITGA6, ITGB4 |
Laminina 5 Kolagen XVII, laminina 5 α6 β4 Integryna |
OMIM 226700 |
| EB dystrophica (EBD) |
EBD Hallopeau-Siemens EBD non-Hallopeau-Siemens EBD dziedziczona autosomalnie dominująco |
COL7A1 COL7A1 COL7A1 |
Kolagen VII Kolagen VII Kolagen VII |
OMIM 226600 OMIM OMIM 131800 |
EB epidermolytica (EB simplex, EBS)
W tej grupie wyróżnia się:
- odmianę poronną Webera-Cockayne’a postaci zwykłej EB (EBS-WC) OMIM 131800
- odmianę Koebnera postaci zwykłej EB (EBS-K, EBS2) OMIM 131900
- odmianę Dowlinga-Meary postaci zwykłej EB (EBS-DM) OMIM 131760
- EBS z cętkowaną pigmentacją (EB with mottled pigmentation; EBS-MP) OMIM 131960
- odmianę z dystrofią mięśniową postaci zwykłej EB (EB simplex and limb-girdle muscular dystrophy MD-EBS; MDEBS, EB-MD) OMIM 226670
- odmiana Ogna (EBSO, EBS1) OMIM 131950
- EB simplex dziedziczona autosomalnie recesywnie OMIM 601001
EB junctionalis (EBJ)
- odmiana Herlitza (letalna) EBJ (odmiana Herlitza-Pearsona, epidermolysis bullosa letalis, EBJ) OMIM 226700
- odmiana ‘non-Herlitz’ EBJ (EB, generalized atrophic benign, GABEB) OMIM 226650
- odmiana EBJ z atrezją odźwiernika, zespół Carmiego (EB with pyloric atresia, EB-PA, PA-JEB) OMIM 226730
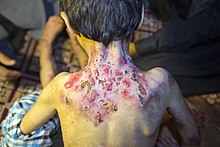
Postać dystroficzna (EB dystrophica)
W obrębie tej podgrupy wyróżnia się następujące jednostki chorobowe:
- postać dystroficzna EB odmiana Hallopeau-Siemensa (EB dystrophica, Hallopeau-Siemens type; severe RDEB); EBR1 OMIM 226600
- postać dystroficzna EB odmiana ‘non-Hallopeau-Siemens’ (RDEB mitis)
- postać dystroficzna EB odmiana dziedziczona autosomalnie dominująco
- postać dystroficzna wariant Pasiniego (EB dystrophica, Pasini type, allopoliploid dominant dystrophic EB; EBDD OMIM 131750)
- postać dystroficzna wariant Cockayne’a-Toureine’a (EB dystrophica, Cockayne-Touraine type) OMIM 131800
- postać Barta (EB with congenital localized absence of skin and deformity of nails, EB dystrophica, Bart type) OMIM 132000
Charakterystyka poszczególnych odmian EB
Grupa epidermolysis bullosa simplex
EB Webera-Cockayne’a
Choroba spowodowana jest mutacją w genie KRT14 kodującego keratynę 5 i 14 oraz defektem enzymów proteolitycznych. Dziedziczona jest autosomalnie dominująco. Zmiany występują najczęściej na stopach, rzadziej na dłoniach; mogą jednak pojawić się w każdej lokalizacji. Przebieg jest stosunkowo łagodny. Pęcherze rzadko obecne są już przy urodzeniu; zwykle pojawiają się u dzieci w wieku około 18 miesięcy, gdy wykazują dużą aktywność ruchową. Zmiany mogą się ujawnić w późniejszym wieku, w późnym dzieciństwie albo u młodych dorosłych. Klasyczne są przypadki młodych rekrutów u których choroba ujawniła się po pierwszym forsownym marszu. Czynniki nasilające tworzenie pęcherzy to duża wilgotność, wzmożone pocenie się. Błony śluzowe i płytki paznokciowe nie są zajęte.
EB Dowlinga-Meary (EB herpetiformis)
Od innych chorób z grupy epidermolysis bullosa simplex różni się ciężkim przebiegiem i charakterystycznym obrazem w mikroskopie elektronowym (cytoplazmatyczne „kępki” tonofilamentów). Tak jak typ Koebnera i typ Webera-Cockayne’a, typ Dowlinga-Meary EBS wiąże się z mutacjami w genach KRT5 (region 12q13.13) i KRT14 (region 17q21.2). Objawy: uogólnione tworzenie pęcherzy o opryszczkowatym (zgrupowanym) układzie, stąd inna nazwa schorzenia, epidermolysis bullosa simplex herpetiformis. Surowicze i krwotoczne pęcherze tworzą się w każdej lokalizacji, przede wszystkim jednak na stopach i podeszwach, dookoła ust, na karku i szyi. Bliznowacenie nie jest regułą, ale proces zapalny, zwłaszcza wikłający pęcherze krwotoczne, może skutkować tworzeniem blizn i prosaków. Notowano zajęcie błon śluzowych i płytkowatą hiperkeratozę dłoni i podeszew. Podpaznokciowe tworzenie pęcherzy może powodować utratę paznokci. Czynnikami zaostrzającymi przebieg choroby są: gorączka, pora roku (miesiące letnie), a także wiek (zmiany nasilają się z wiekiem).
EB wariant Koebnera
Choroba spowodowana jest mutacjami w genach keratyny 5 i 14 (KRT4, KRT15). Ponieważ mutacje w tych samych genach odpowiadają za wystąpienie cech fenotypowych EBS typu Webera-Cockayne’a i typu Dowlinga-Meary, często cechy tych trzech chorób nakładają się na siebie. Zdarza się, że w obrębie jednej rodziny diagnozowano przypadki dwóch różnych typów EBS. Zmiany są hiperkeratotyczne, pęcherzowe, czasem pęcherze są krwotoczne; w obrazie klinicznym występują nadżerki. Manifestują się już od urodzenia albo we wczesnym niemowlęctwie. Niekiedy tworzą się prosaki, mogą wystąpić zaburzenia pigmentacji (hipo- lub hiperpigmentacja) tak jak w odmianie Dowlinga-Meary, czasem zajęte są paznokcie i błony śluzowe.
Grupa epidermolysis bullosa junctionalis
EB Herlitza
Postać śmiertelna, rzadka. Dziedziczona jest autosomalnie recesywnie. Spowodowana jest mutacją w jednym z genów kodujących trzy polipeptydy lamininy 5: α-3 (gen LAMA3), β-3 (gen LAMB3), albo γ-2 (gen LAMC2). U noworodków od urodzenia występują liczne pęcherze, które łatwo pękają, pozostawiając rozległe nadżerki w obrębie całej skóry i błon śluzowych, również dróg oddechowych. Już wewnątrzmacicznie następuje tworzenie pęcherzy i nadżerek, tak że dziecko rodzi się z rozległymi ubytkami naskórka. Ręce i stopy są zwykle relatywnie najmniej dotknięte przez proces chorobowy. Nigdy nie występuje bliznowacenie, syndaktylia, nie tworzą się prosaki. W wieku około 6 miesięcy wokół ust bądź nosa tworzy się patognomoniczny, niegojący się strup. Zgon następuje w okresie niemowlęcym mimo intensywnej opieki medycznej, stosowania antybiotyków, kortykosteroidów, wyrównania poziomu elektrolitów. Przyczyną śmierci jest utrata białka surowicy i sepsa. W ostatnich latach podejmowano próby przeszczepiania skóry.
EB z atrezją odźwiernika
Bardzo rzadka choroba spowodowana mutacją w genach ITGB4 i ITGA6. W skórze chorych można stwierdzić obniżoną ekspresję kodowanych przez te geny α6 i (lub) β4 integryny. Dziedziczenie jest autosomalne recesywne. Objawy są obecne przy urodzeniu albo pojawiają się we wczesnym niemowlęctwie. Zmianom skórnym z reguły towarzyszy wrodzona atrezja odźwiernika (rzadziej atrezja przełyku lub dwunastnicy). Często występuje dystrofia paznokci i anomalie w zakresie uzębienia, takie jak hipoplazja szkliwa. Tworzenie pęcherzy jest uogólnione, rokowanie bardzo złe, mimo chirurgicznej interwencji; mutacja często jest letalna. W wariantach letalnych (zwykle spowodowanych mutacją typu nonsens) zgon następuje wkrótce po urodzeniu; w pozostałych przypadkach obserwuje się tendencję do zaostrzania zmian skórnych z wiekiem.
Grupa epidermolysis bullosa dystrophica
EB dystroficzna, postać Pasiniego
Dziedziczona jest w sposób autosomalny dominujący. Pęcherze ustępują z pozostawieniem drobnych, białawych wykwitów grudkowych, umiejscowionych głównie na tułowiu w okolicy krzyżowej. Zajęte są śluzówka jamy ustnej i zęby. Paznokcie mogą być dystroficzne. W starszym wieku choroba może złagodzić swój przebieg.
EB dystroficzna, postać Cockayne’a-Toureine’a
Dziedziczona jest w sposób autosomalny dominujący. Wykwity głównie na kończynach, śluzówka i zęby zajęte w minimalnym stopniu.
EB Hallopeau-Siemensa
Choroba wywołana jest mutacją w genie kodującym kolagen typu VII (COL7A1). Charakterystyczne jest uogólnione tworzenie pęcherzy w momencie urodzenia lub we wczesnym niemowlęctwie, rozległe bliznowacenie, zmiany w obrębie skóry kończyn. Miejsca mające predylekcję do pęcherzy to dłonie, stopy, łokcie i kolana. Może wystąpić pseudosyndaktylia dłoni i stóp, niekiedy są zajęte paznokcie i zęby, śluzówka przełyku (czego skutkiem jest zwężenie przełyku), stenoza cewki moczowej, odbytu, stulejka, bliznowacenie rogówki lub spojówki. W EB Hallopeau-Siemensa wyraźnie zwiększone jest ryzyko wystąpienia raka kolczystokomórkowego skóry; nowotwór rozwija się na podłożu blizn.
Bibliografia
- Stefania Jabłońska, Sławomir Majewski Choroby skóry i choroby przenoszone drogą płciową PZWL 2005, ISBN 83-200-3367-5.
- Gernot Rassner, Dermatologia. Podręcznik i atlas, Wojciech Silny (tłum.), Wrocław: Urban & Partner, 1994, ISBN 83-85842-25-X, ISBN 3-541-08374-3, OCLC 749381926.

Linki zewnętrzne
- Epidermolysis bullosa with pyloric atresia. gfmer.ch. [zarchiwizowane z tego adresu (2006-12-15)]. na stronie Geneva Foundation for Medical Education and Research (ang.)
- Artykuł z eMedicine (ang.)
- Epidermolysis Bullosa Medical Research Foundation. med.stanford.edu. [zarchiwizowane z tego adresu (2006-12-31)]. (ang.).
- Netzwerk Epidermolysis bullosa. netzwerk-eb.de. [zarchiwizowane z tego adresu (2007-02-18)]. (niem.).
- Epidermolysis Bullosa Simplex. geneclinics.org. [zarchiwizowane z tego adresu (2007-02-20)]. w GeneReviews (ang.)