
| Adenomatous polyposis coli | |
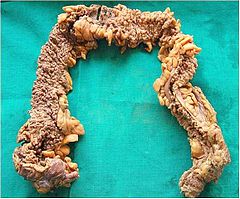 Liczne polipy w preparacie po kolektomii | |
| Klasyfikacje | |
| ICD-10 |
D12 |
|---|---|
| D12.0 |
Jelito ślepe |
| D12.1 |
Wyrostek robaczkowy |
| D12.2 |
Okrężnica wstępująca |
| D12.3 |
Okrężnica poprzeczna |
| D12.4 |
Okrężnica zstępująca |
| D12.5 |
Okrężnica esowata |
| D12.6 |
Okrężnica, nie określona |
| D12.7 |
Zgięcie esiczo-odbytnicze |
| D12.8 |
Odbytnica |
| D12.9 |
Odbyt i kanał odbytu |
| OMIM | |
| MeSH | |
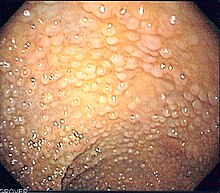
Rodzinna polipowatość gruczolakowata (ang. familial adenomatous polyposis, FAP) – choroba genetyczna charakteryzująca się licznymi polipami gruczolakowatymi jelita grubego, powstającymi w 2. dekadzie życia. U nieleczonych chorych niemal we wszystkich przypadkach rozwija się rak jelita grubego.
Epidemiologia
Rodzinna polipowatość gruczolakowata to rzadka choroba. W 2009 roku Europejska Agencja Leków (EMEA, ang. European Medicines Agency) oszacowała jej częstość w populacji Unii Europejskiej na 3-10/100 000. Jest traktowana jako stan przedrakowy, który nieuchronnie prowadzi do powstania raka jelita grubego. Zwykle nie ujawnia się przed 10. rokiem życia. Jej pełny obraz powstaje zwykle w 3. – 4. dekadzie życia. Tylko wyjątkowo objawy mogą się pojawić w okresie noworodkowym lub wczesnoniemowlęcym.
Etiologia
Choroba jest dziedziczona autosomalnie dominująco. FAP spowodowana jest mutacjami antyonkogenu APC (adenomatous poliposis coli gene) w locus 5q21-22. Mutacje te prowadzą do powstania nieprawidłowych, skróconych, białek. W około 25-30% przypadków mutacja ma charakter spontaniczny i u tych chorych wywiad rodzinny jest ujemny.
Patomorfologia
Charakterystyczne są polipy gruczolakowate, zarówno cewkowe, kosmkowe jak i mieszane. Umiejscowione są na całej długości jelita, ze szczególnym uwzględnieniem esicy i odbytnicy. Z reguły nie przekraczają 1 cm. Makroskopowo mogą przypominać powiększone grudki chłonne okrężnicy, co może być przyczyną nierozpoznania choroby w badaniu kolonoskopowym.
Objawy i przebieg

Głównym objawem są setki polipów gruczolakowatych różnej wielkości w okrężnicy i odbytnicy. U połowy pacjentów z FAP polipy są obecne w wieku 15 lat, a w wieku 35 lat u 95%. Oprócz tego może współistnieć polipowatość dna żołądka (ang. fundic gland polyps, FGP), polipy dwunastnicy i gruczolaki jelita cienkiego.
Na podłożu gruczolaków rozwijają się nowotwory złośliwe. Najczęstszym nowotworem w zespole polipowatości gruczolakowatej jest rak jelita grubego, którego ryzyko wynosi niemal 100%. U nieleczonych chorych rak jelita grubego powstaje przeciętnie w wieku 35 lat, a rzadko przed 20. rokiem życia.
Zespół powoduje powstawanie również innych nowotworów, z których najczęstszymi są rak brodawkowaty tarczycy i gruczolakorak jelita cienkiego.
Nowotwory złośliwe związane z FAP:
- rak tarczycy – najczęściej jest to rak brodawkowaty tarczycy, inne typy nowotworów złośliwych tarczycy są bardzo rzadkie, nowotwór w zespole FAP może występować z częstością od 0,6% do 6,1%. Zalecany jest stały nadzór na chorymi.
- gruczolakorak jelita cienkiego – mnogie gruczolaki występują u 50-90% chorych, a u 3-5% chorych może rozwinąć się gruczolakorak. Ryzyko nowotworu jest zwiększone 300-krotnie.
- hepatoblastoma – u dzieci z zespołem FAP występuje aż 750-7500 razy częściej niż w populacji ogólnej. Mimo że skuteczność badań przesiewowych jest niejasna, niektórzy autorzy zalecają nadzór nad dziećmi z potwierdzonym zespołem.
- rak trzustki – zespół powoduje 4,5-krotny wzrost ryzyka zachorowania na ten typ nowotworu.
- rak żołądka – mimo że polipy w żołądku występują u połowy chorych, to ryzyko raka żołądka w krajach zachodnich jest bardzo niskie, znacznie wyższe występuje w krajach Azjatyckich, gdzie ryzyko może być nawet 10-krotnie wyższe niż w populacji ogólnej.
- rak nadnerczy – gruczolaki nadnerczy występują 4 razy częściej niż w populacji ogólnej, jednak przebieg kliniczny nie odbiega od przypadków nie uwarunkowanych zespołem, rak nadnerczy w zespole FAP występuje rzadko.
- guzy mózgu – najczęstszym guzem OUN związany z FAP jest rdzeniak, stanowi około 80% guzów związanych z zespołem FAP, ryzyko zachorowania w ciągu życia wynosi około 1%.
Choroba może dawać też zróżnicowane objawy pozajelitowe, takie jak:
- kostniaki – występują u 20% chorych, zwykle w obrębie żuchwy i czaszki.
- nieprawidłowości uzębienia – występują u 17% chorych, nieprawidłowości obejmują zęby nadliczbowe, wrodzone braki zębów, torbiele zębowe, zębiaki,
- guzy desmoidalne – miejscowo złośliwy guz najczęściej zlokalizowany w jamie brzusznej, tylko 10% z nich znajduje się poza jamą brzuszną. Często rosną do ogromnych rozmiarów, stanowią duże zagrożenie dla sąsiednich struktur,
- torbiele epidermoidalne – nie wykazują potencjału złośliwego, stanowią problem kosmetyczny,
- wrodzona hipertrofia barwnikowa siatkówki.
Zespół Gardnera
Odmianą FAP jest zespół Gardnera, gdzie występowaniu setek, a nawet tysięcy polipów jelita grubego towarzyszy powstawanie guzów mezodermalnych o typie kostniaków, guzów desmoidalnych, torbieli naskórkowych oraz przerostu nabłonka barwnikowego siatkówki (CHRPE, ang. Congenital Hypertrophy of the Retinal Pigment Epithelium).
Zespół Turcota
Współwystępowanie guzów mózgu i polipów okrężnicy nazywamy zespołem Turcota.
Poronna postać FAP
Mniej agresywny wariant FAP określa się jako poronny (attenuated FAP, AFAP). W jego przebiegu powstaje mniej polipów (10-100), a polipy rozwijają się w późniejszym wieku. Ryzyko nowotworu złośliwego jest mniejsze i nie przekracza 70%.
Rozpoznanie
Rozpoznanie FAP stawiane jest na podstawie badania kolonoskopowego.
Leczenie
Postępowaniem z wyboru jest leczenie operacyjne – najczęściej całkowita kolektomia. Istnieją próby leczenia profilaktycznego w postaci podawania niesteroidowych leków przeciwzapalnych, np. celekoksybu, sulindaku.