
| syndroma Marfan | |
| |
| Klasyfikacje | |
| ICD-10 | |
|---|---|

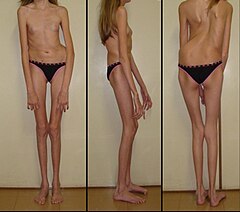
Zespół Marfana (ang. Marfan syndrome, MFS) – choroba genetyczna tkanki łącznej z grupy fibrylinopatii, charakteryzująca się dużą zmiennością fenotypową. Przyczyną zespołu jest mutacja w genie fibryliny-1 (FBN1). Mutacja w około 25% występuje de novo (nieodziedziczona po rodzicach). Zmiany narządowe w przebiegu zespołu Marfana dotyczą całego organizmu, niemniej najbardziej charakterystyczne to te związane z narządem wzroku, układu ruchu, serca i naczyń krwionośnych. Częstość zespołu szacowana jest na 1–2:10 000.
Historia
Pierwszego typowego opisu tej choroby dokonał w 1876 roku okulista z Cincinnati w USA E. Williams. W 1896 pediatra z Paryża, dr Antoine Bernhard-Jean Marfan opisał zespół nieprawidłowości szkieletowych u 5½-letniej dziewczynki Gabrielle P. i wprowadził na ich określenie pojęcie dolichostenomelii. Ta sama pacjentka została przebadana ponownie w wieku 11½ lat i opisano wtedy silną skoliozę i przykurcze palców bez nieprawidłowości ocznych i naczyniowych. W 1902 roku Francuz A. Achard wprowadził do literatury termin arachnodaktylii, oznaczający pająkowatość palców (ponad proporcję długie i cienkie palce dłoni). W 1914 Niemiec Friedrich Börger jako pierwszy opisał u pacjentów z arachnodaktylią podwichnięcie soczewek. Dziedziczenie autosomalne dominujące zespołu opisano w 1931 roku.
Eponimu „choroby Marfana” użył przypuszczalnie jako pierwszy Francuz A. Carrau w 1929 roku. Wcześniej zespół określano jako arachnodaktylię.
W 1943 pojawiły się doniesienia o tętniaku rozwarstwiającym aortę w jej części wstępującej u pacjentów z zespołem Marfana. W 1950 dokonano analizy rodowodów wielu pacjentów, głównie z powikłaniami sercowo-naczyniowymi, co pozwoliło określić różnorodność i zmienność obrazu chorobowego. W 1956 określono podobieństwo strukturalne między więzadełkami soczewek oczu a warstwą środkową aorty. Beals i Hecht w 1971 opisali chorobę dziedziczoną autosomalnie dominująco, którą nazwali CCA (ang. congenital constractural arachnodactyly – wrodzona arachnodaktylia, pająkowatość palców, z przykurczem), obecnie znana jako zespół Bealsa albo zespół Bealsa-Hechta. Zmiany szkieletowe w tej chorobie odpowiadają tym, jakie opisał dr Antoine Marfan, zaś oczy i aorta są zdrowe lub mają mniejsze wady, występuje znacznie mniejsza ruchomość więzadeł oraz przykurcze.
W 1986 roku odkryto fibrylinę; glikoproteinę, której wadliwa budowa leży u podłoża zespołu Marfana. Hollister i wsp. w 1991 odkryli geny odpowiedzialne za budowę fibryliny w locus (obszarze chromosomu) 15q 15-21. W tym samym roku Lee i wsp. zlokalizowali na chromosomie 5 (locus 5q 23–31) zespół genów także odpowiedzialny za syntezę fibryliny, a zmieniony chorobowo w postaci CCA.
Etiologia

Przyczyną zespołu Marfana jest uwarunkowane genetycznie uszkodzenie włókien sprężystych i zaburzenie w tworzeniu elastyny oraz substancji podstawowej tkanki łącznej. Choroba ta dziedziczy się w sposób autosomalny dominujący, charakteryzuje się dużym stopniem penetracji genu oraz różnorodną ekspresją. Gen, którego mutacje są odpowiedzialne za występowanie typowych dla tego zespołu objawów został zlokalizowany na chromosomie 15. Koduje on białko fibrylinę 1. Fibrylina 1 decyduje o prawidłowym wiązaniu białka TGF-β w tkance łącznej. W prawidłowym stanie TGF-β jest przechwytywane przez fibrylinę do tkanki łącznej. W zespole Marfana białko TGF-β nie jest wiązane przez fibrylinę i pozostaje jako wolna cząstka we krwi, co prowadzi do nieprawidłowych zachowań komórek tkanki łącznej.
Mutacje fibryliny-1 stwierdzono w innych chorobach genetycznych, określanych niekiedy jako typu I. Są to:
- dziedziczone autosomalnie zwichnięcie soczewki
- zespół Shprintzena-Goldberga
- zespół Weilla-Marchesaniego
- zwichnięcie soczewki
- rodzinny tętniak aorty
- fenotyp MASS
- nowy wariant zespołu Marfana
- marfanoidalne nieprawidłowości kostne
- zespół Acharda
- zespół Bealsa
- zespół Ehlersa-Danlosa (EDS)
- zespół Loeysa-Dietza.
Obraz kliniczny
- wysoki wzrost:
- średnia długość ciała noworodka 53 ± 4,4 cm dla chłopców, 52,5 ± 3,5 cm dla dziewczynek
- średni ostateczny wzrost 191,3 ± 9 cm dla mężczyzn, 175,4 ± 8,2 cm dla kobiet
- dysproporcja budowy: stosunek górnej do dolnej części ciała mniejszy niż 0,85
- stosunek zasięgu ramion do wzrostu > 1,05
-
cechy dysmorficzne:
- dolichocefalia
- arachnodaktylia (długie i cienkie palce dłoni)
- hipoplazja (słabe wykształcenie się) policzków
- mikrognacja
- retrognacja
- wysokie i wąskie podniebienie
- stłoczenie zębów; konieczne usuwanie niektórych zębów stałych
- wady narządu wzroku (występują u przynajmniej 60% chorych):
- enoftalmia
- zwichnięcie soczewki (typowe dla zespołu i najczęściej diagnozowane)
- krótkowzroczność
- astygmatyzm
- zwiększony wymiar gałki ocznej w osi długiej
- płaska rogówka
- jaskra (jaskra otwartego kąta przesączania występuje znacząco częściej w każdej grupie wiekowej u osób z zespołem w porównaniu do ogółu populacji, a częstotliwość jej występowania rośnie wraz z wiekiem)
- hipoplazja (słabe wykształcenie się) tęczówki
- odwarstwienie siatkówki (może pojawiać się spontanicznie w oczach z krótkowzrocznością osiową lub w okresie po usunięciu zaćmy, szczególnie w gałkach dłuższych)
- zaćma (rozwija się wcześniej niż w reszcie populacji)
- antymongoloidalne ustawienie szpar powiekowych
- wady układu nerwowego
- wzmożone zapotrzebowanie na sen
- zawroty głowy
- omdlenia
- przewlekłe zmęczenie
- bóle głowy
- zaburzenia koordynacji
- wady układu sercowo-naczyniowego
- niedomykalność zastawki aorty (leczona m.in. metodą Bentalla)
- niedomykalność zastawki mitralnej
- wypadanie płatka zastawki mitralnej
- niedociśnienie tętnicze
- zastoinowa niewydolność serca
- wypadanie płatka zastawki trójdzielnej
- epizodyczna tachykardia nadkomorowa lub bradykardia
- przedwczesne wapnienie pierścienia zastawki mitralnej
- poszerzenie pnia aorty
- nieprawidłowe przewodzenie w węźle zatokowo-przedsionkowym
- tętniak rozwarstwiający aorty
- tętniak aorty wstępującej, rzadziej w innych odcinkach. Rozszerzenie światła aorty wstępującej operuje się chirurgicznie, gdy osiągnie wielkość ok. 50 mm.
- poszerzenie pnia płucnego
- odma opłucnowa
-
rozedma płuc
- duszność
- wady układu pokarmowego:
- zaburzenia wchłaniania pokarmu z żołądka/jelita cienkiego
- nieprawidłowa perystaltyka jelit
- zapalenia/owrzodzenia śluzówki żołądka
- niewydolność wpustu żołądka
- wady układu kostnego:
- klatka piersiowa szewska
- klatka piersiowa kurza
- asymetria mostka i klatki piersiowej
- przepukliny
- przedwczesne zwyrodnienie stawów
-
skolioza
- kifoskolioza
- nasilona lordoza lędźwiowa
- kręgozmyk
- lumbosacral dural ectasia
- protruzja panewek stawów biodrowych
- dolichostenomelia
- nadmierna ruchomość stawów
- przykurcze stawowe
- przeprost stawu kolanowego (genu recurvatum)
- arachnodaktylia
- płaskostopie
- długie, wąskie stopy
- stopa wydrążona
- młotkowate palce stóp
- rotacja przyśrodkowa kostki przyśrodkowej
- obniżona masa mięśniowa
- rozstępy skórne
- zmniejszona ilość tkanki tłuszczowej
W piśmiennictwie notowane jest etniczne zróżnicowanie częstości występowania objawów zespołu pomiędzy Azjatami i pacjentami z Zachodu. Akutsu i wsp. wykazali, że japońscy pacjenci z zespołem Marfana w porównaniu do pacjentów z Zachodu rzadziej mają objawy związane z układem kostnym. Wśród koreańskich pacjentów z Zespołem Marfana notowano rzadsze w stosunku do pacjentów z Zachodu występowanie skoliozy oraz niższy wzrost i niższą masę ciała. W kilku badaniach wykazano także, że objawy dotyczące układu sercowo-naczyniowego mogą częściej występować u Azjatów niż osób z Zachodu.
W badaniu tajwańskim (2017) stwierdzono w grupie pacjentów z zespołem Marfana podwyższone ryzyko rozwoju różnego typu nowotworów.
Rozpoznanie
Diagnostyka zespołu Marfana opiera się na obecności charakterystycznych odchyleń od stanu prawidłowego stwierdzanych w badaniu klinicznym w obrębie układu kostnego, naczyniowego oraz narządu wzroku. Dodatkowym czynnikiem ułatwiającym znacznie ustalenie rozpoznania jest dokładnie przeprowadzony wywiad rodzinny. Rozpoznanie oparte na badaniu klinicznym i wywiadzie rodzinnym, ustala się na podstawie obecności typowych objawów współistniejących w 3 układach przy negatywnym wywiadzie rodzinnym (jeśli rodzice chorego są zdrowi), oraz w 2 lub więcej układach przy stwierdzeniu obciążonego wywiadu rodzinnego. Objawy charakterystyczne dla tego zespołu mogą być stwierdzane już przy urodzeniu, chociaż nie jest to regułą.
U większości chorych zespół Marfana można rozpoznać już u noworodka, który jest smukły i nie ma właściwego napięcia mięśniowego. W późniejszym okresie można stwierdzić, że dziecko niedowidzi, gdyż nie śledzi wzrokiem poruszających się przedmiotów. Innym bardzo ważnym, często występującym i łatwym do wykrycia przez pediatrę objawem jest słyszalny, charakterystyczny szmer w sercu. Czasami jednak wada serca ujawnia się dopiero u starszego dziecka, np. dwuletniego czy pięcioletniego (czasami jeszcze później), co też jest dla tego zespołu typowe. Dziecko rośnie szybciej od zdrowych rówieśników, jest bardzo smukłe i ma trudności w siadaniu, a potem szybkim bieganiu. Przyczyną tych problemów małego dziecka (do 3 lat) jest jego wiotki układ mięśniowy.
Czasem dziecko ma niebieskawe twardówki, ślini się nawet do czwartego roku życia z powodu wiotkości mięśni mimicznych twarzy. Palce dziecka są smukłe i zdarza się, że połówki ciała dziecka są asymetryczne np. jedna ręka jest grubsza i dłuższa od drugiej, albo jedna noga jest grubsza i dłuższa od drugiej). Ta asymetria ciała może być przyczyną bocznego skrzywienia kręgosłupa, który z przyczyn typowych dla zespołu Marfana jest słaby i wiotki. Mimo często bardzo słabego wzroku poziom umysłowy dziecka nie odbiega zwykle od normy. Z powodu słabości mięśni klatki piersiowej, a także skrzywienia bocznego kręgosłupa – zwłaszcza jeżeli jest duże – upośledzone jest oddychanie dziecka. Dlatego też dzieci z zespołem Marfana często chorują na zapalenie oskrzeli, płuc, a wielu dorosłych cierpi na astmę.
Prowadzone są badania naukowe mające na celu określenie ilościowych cech zespołu Marfana w wyglądzie twarzy za pomocą pomiarów morfometrycznych na podstawie przestrzennych odwzorowań twarzy wykorzystujących obrazowanie trójwymiarowe. Badania te wykazały w pomiarach pewne cechy wspólne w kształtach twarzy chorych na zespół.
Różnicowanie
W diagnostyce różnicowej zespołu Marfana należy uwzględnić:
- homocystynurię
- zespół Sticklera
- zespół Ehlersa-Danlosa
- zespół Bealsa
- dystrofię miotoniczną
- zespół Sotosa
- marfanoidalny zespół nadmiernej ruchomości stawów (Marfanoid hypermobility syndrome)
- zespół Klinefeltera
- niedokrwistość sierpowatokrwinkowa
- MEN2B
- rodzinne wypadanie płatka zastawki mitralnej
- opóźnienie umysłowe z cechami marafnoidalnymi sprzężone z chromosomem X
- rodzinne zwichnięcie soczewki
- zespół Weilla-Marchesaniego
- rodzinna dwupłatkowa zastawka mitralna
Rokowanie
U schyłku XIX i do lat 60. XX wieku chorzy z zespołem Marfana umierali w młodości (do 30. roku życia). Obecnie istnieją znacznie większe możliwości kontrolowania powikłań narządowych zespołu Marfana i średni czas przeżycia jest nieco dłuższy. Na początku XXI wieku śmiertelność wśród osób z Zespołem Marfana jest 20-40-krotnie większa niż u zdrowych osób w tym samym wieku. Najczęstszą przyczyną zgonów w Zespole Marfana jest rozwarstwienie pnia aorty. W celu ograniczenia powikłań związanych z aortą stosuje się atenolol lub losartan, które według metaanalizy z 2016 roku są równorzędnie skuteczne (nie stwierdzono, by jeden lek miał przewagi nad drugim).
Ze względu na zmiany w obrębie aorty zaleca się, aby pacjenci z zespołem Marfana powstrzymywali się od intensywnego wysiłku fizycznego, aby zapobiec zwiększaniu ciśnienia krwi podczas ćwiczeń (co stwarza ryzyko poszerzenia tętnicy aorty i pęknięcia). Mas-Stachurska i in. w badaniu na myszach (2017) doszła do wniosku, że umiarkowany, długotrwały wysiłek fizyczny jest jednak korzystny, ponieważ łagodzi rozwój fenotypu sercowo-naczyniowego w zespole (ogranicza rozwój tętniaka).
Znani chorzy
Sugerowano lub potwierdzono, że zespół Marfana mieli lub mają:
- Isaiah Austin
- Austin Carlile
- Echnaton
- Robert Johnson
- Abraham Lincoln
- Peter Mayhew
- Niccolò Paganini
- Siergiej Rachmaninow
- Joey Ramone
- Vincent Schiavelli
- Troye Sivan
- Jonathan Larson
- Javier Botet
Bibliografia
- Harold Chen: Atlas of Genetic Diagnosis and Counselling. Totowa, NJ: Humana Press, 2006, s. 619-629. ISBN 1-59259-956-7.
Linki zewnętrzne
- Mafran Syndrome; MFS w bazie Online Mendelian Inheritance in Man (ang.)
- polskie Stowarzyszenie Rodzin Chorych na Zespół Marfana (pol.)
- National Marfan Fundation – fundacja na rzecz badań, profilaktyki i leczenia zespołu Marfana (USA), marfan.org [zarchiwizowane z adresu 2013-09-14] (ang.).
- brytyjskie stowarzyszenie osób z Zespołem Marfana (Marfan Association UK) (ang.)
- Amerykańskie centrum badań nad zespołem Marfana działające w szkole medycyny Uniwersytetu Johnsa Hopkinsa, hopkinsmedicine.org [zarchiwizowane z adresu 2013-12-06].