
| Syndroma Clouston | |
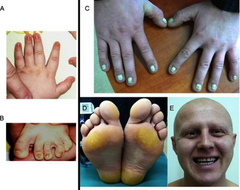 Fenotyp dwóch pacjentów z zespołem Cloustona: Pacjent nr 1: A – dysplastyczne paznokcie u rąk, B – dysplastyczne paznokcie u stóp, Pacjent nr 2: C – dysplastyczne oraz pogrubiałe paznokcie u rąk, D – rogowiec stóp, E – częściowe łysienie, rzadkie brwi i rzęsy, normalne zęby | |
| Klasyfikacje | |
| ICD-10 |
Q82.8 |
|---|---|
| OMIM | |
| MeSH | |
Zespół Cloustona, in. dysplazja ektodermalna z zachowaną czynnością gruczołów potowych (ang. Clouston’s hidrotic ectodermal dysplasia, alopecia congenita with keratosis palmoplantaris, Clouston syndrome, Fischer–Jacobsen–Clouston syndrome, Waldeyer-Fisher syndrome, hidrotic ectodermal dysplasia, keratosis palmaris with drumstick fingers, palmoplantar keratoderma and clubbing) – genetycznie uwarunkowany zespół wad wrodzonych, dziedziczony autosomalnie dominująco. Charakteryzuje się triadą objawów, na którą składają się: dysplazja paznokci, utrata włosów oraz rogowiec dłoni i stóp, bez objawów niepełnosprawności intelektualnej.
Zespół został po raz pierwszy opisany w 1929 przez kanadyjskiego lekarza Howarda Cloustona.
Etiologia
Choroba spowodowana jest uszkodzeniem genu GJB6 zlokalizowanego na ramieniu długim chromosomu 13, w regionie q12. Mutacja genu powoduje uszkodzenie białka koneksyny-30.
Epidemiologia
Częstość występowania szacowana jest na 1–9 na 100 000 urodzeń. Zespół jest przykładem genetycznego efektu założyciela w populacji Frankokanadyjczyków. W Polsce została opisana jedna rodzina z tym zespołem (2015).
Obraz kliniczny
Zespół Cloustona charakteryzuje się triadą objawów, na którą składają się dysplazja paznokci, utrata włosów oraz rogowiec dłoni i stóp. Najbardziej charakterystycznym klinicznie oraz najczęściej występującym objawem jest dysplazja paznokci, która jest widoczna już przy urodzeniu lub pojawia się w okresie niemowlęcym. Paznokcie są pogrubione, wolno rosnące, łamliwe, często nadmiernie wypukłe, przebarwione oraz prążkowane, może dochodzić do onycholizy oraz nawracających zakażeń i całkowitej utraty paznokci. Utrata włosów może być widoczna już przy urodzeniu lub pojawiać się najpóźniej w okresie dziecięcym, całkowity brak włosów najczęściej obejmuje owłosienie głowy, pach oraz owłosienie łonowe, jednakże może być uogólniony. Rogowiec dłoni i stóp nie jest stałym objawem zespołu, pojawia się zwykle w dzieciństwie, z wiekiem następuje zarówno narastanie zmian na dłoniach i stopach, jak i mogą się pojawiać się ogniska rogowacenia w innych miejscach niż dłonie i stopy (transgrediencja). Zwykle nie występują wady w zakresie uzębienia i zawsze zachowana jest zdolność wydzielania potu. Zespół nie powoduje niepełnosprawności intelektualnej.
Diagnostyka różnicowa
Zespół Cloustona należy różnicować z zespołem Coffina-Siris, zespołem dysplazji ektodermalnej Rappa-Hodgkina oraz wrodzonym zgrubieniem paznokci.
Ostatecznym potwierdzeniem rozpoznania jest potwierdzenie specyficznej mutacji w genie GJB6
Leczenie i rokowanie
Nie ma specyficznego leczenia zespołu Cloustona (2016). Rokowanie co do życia jest dobre, długość życia również nie jest skrócona.
Linki zewnętrzne
- OMIM Entry – # 129500 – CLOUSTON SYNDROME, www.omim.org [dostęp 2016-12-30].