
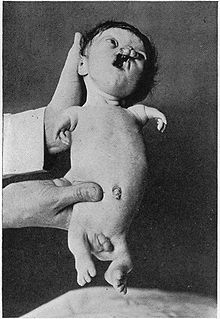


Zespół Robertsa (zespół pseudotalidomidowy, zespół Appelta-Gerkena-Lenza, ang. Roberts' syndrome, pseudothalidomide syndrome, Appelt-Gerken-Lenz syndrome) – rzadki, uwarunkowany genetycznie zespół wad wrodzonych, charakteryzujący się symetrycznym skróceniem wszystkich kończyn i niezwykłą nieprawidłowością cytogenetyczną, jaką jest przedwczesne oddzielenie centromeru, uniemożliwiające proces parowania chromatyd.
Historia
Nazwa zespołu upamiętnia Johna Binghama Robertsa, który opisał przypadek zespołu w 1919 roku. Roberts w krótkim doniesieniu zaakcentował fakt pokrewieństwa rodziców, ale nie określił sposobu dziedziczenia.
Z czasem w literaturze znaleziono szereg znacznie wcześniejszych przekazów o urodzeniach dzieci, spełniających kryteria zespołu Robertsa. S. Batman mógł opisać chorobę w 1581 roku, jednak jego opis jest zbyt niedokładny. Francois Bouchard opisał zespół, jak się wydaje, najwcześniej, bo w 1672 roku. Inny opis autorstwa niemieckiego lekarza Gottlieba Fridericiego pochodzi z 1737 roku. W XIX wieku przypadki domniemanego zespołu Robertsa przedstawili Mayer w 1829 roku i Isidore Geoffrey St-Hilaire w 1838; wprowadził on do medycyny pojęcie fokomelii, wywiedzione z greckiego phoka oznaczającego fokę. Rudolf Virchow w 1898 roku opisał przypadek zmarłego po porodzie dziecka, również spełniającego kryteria zespołu.
Inne wczesne opisy były autorstwa Stroera oraz Appelta i wsp.. Appelt i wsp. jako pierwsi uznali, że symetryczna fokomelia opisana przez Robertsa stanowi odrębny zespół wad wrodzonych. Appelt kierował w tym czasie pediatryczną sekcją Kreiskrankenhaus Eisenach w Niemczech Wschodnich, i potajemnie opublikował pracę razem z dwoma kolegami z Niemiec Zachodnich. Herrmann, Opitz i wsp. w 1969 roku opisali zespół, który nazwali zespołem SC (od pierwszych liter nazwisk rodzin probandów), Temtamy w 1974 roku określiła ten zespół jako tożsamy z zespołem Robertsa.
Etiologia
Zespół Robertsa spowodowany jest mutacjami w genie ESCO2 (cohesion 1 homolog 2 gene) w locus 8p21.1, którego produkt białkowy odpowiada za prawidłowe tworzenie się par chromatyd siostrzanych w fazie S cyklu komórkowego.
Objawy i przebieg
- malformacje czaszkowo-twarzowe (bardzo duża zmienność):
- obustronne rozszczepy wargi i podniebienia w ciężkich przypadkach
- brak rozszczepów wargi lub podniebienia w niektórych przypadkach
- hiperteloryzm oczny
- nieprawidłowości oczne:
- wytrzeszcz spowodowany płytkością oczodołów
- niebieskawe twardówki
- małoocze
- koloboma powieki
- anomalia Petersa
- zmętnienie rogówki
- zaćma
- szeroki grzbiet nosa
- hipoplastyczne skrzydełka nosa
- naczyniaki wargi, nosa, twarzy lub czoła
- mikrognacja
- malformacje małżowin usznych, brak płatka, nisko osadzone, zrotowane do tyłu małżowiny
- ciemne włosy skalpu z czasem cieńczeją i przybierają srebrzystą barwę
- wady kończyn:
- fenotyp bardzo zmienny
- zazwyczaj symetryczne
- zazwyczaj silniej wyrażone w kończynach górnych niż dolnych
- w najcięższej postaci zupełny brak kończyn i szczątkowe palce, w łagodnych niewielkie skrócenie długości kończyn
- fokomelia, charakterystyczna tetrafokomelia (-fokomelia wszystkich czterech kończyn), w 11% ubytki kończyn dotyczą dwóch z nich, brak fokomelii w 2%
- często zmniejszona liczba palców (oligodaktylia)
- dysplazja lub aplazja kości promieniowej
- brak 1. kości śródręcza, kciuka lub 1. paliczka
- inne wady:
- wady ośrodkowego układu nerwowego
- opóźnienie umysłowe
- małogłowie
- krótkogłowie
- wodogłowie
- agenezja opuszek węchowych
- zwapnienia jąder podstawy
- encephalocele
- porażenia nerwów czaszkowych
- drgawki
- wrodzone wady serca
- wady nerek
- nerki wielotorbielowate
- nerki dysplastyczne
- nerka podkowiasta
- wodonercze
- agenezja nerki
- niedrożność przewodu pokarmowego
- śledziony dodatkowe, połączenie śledzionowo-mosznowe
- wnętrostwo
- spodziectwo
- makropenis lub klitoromegalia, przerost warg sromowych mniejszych
- macica dwurożna
- niechęć do ssania
- mięsak groniasty
- czerniak złośliwy.
- wady ośrodkowego układu nerwowego
Różnicowanie
- zespół SC fokomelia, obecnie uważany za ten sam zespół (Roberts-SC phocomelia syndrome): tetrafokomelia, srebrzyste włosy, naczyniak twarzy, hipoplastyczne skrzydełka nosa; zazwyczaj brak rozszczepów linii środkowej twarzy, przedłożone przeżycie, mniejszy stopień opóźnienia umysłowego lub fizycznego, zazwyczaj mniej nasilona fokomelia
- zespół TAR: aplazja kości promieniowej, obecne kciuki, trombocytopenia hipomegakariotyczna, brak rozszczepu podniebienia
- embriopatia talidomidowa
- zespół Holt-Orama
- tetrafokomelia Zimmera
- zespół Ballera-Gerolda
Bibliografia
- Harold Chen: Atlas of Genetic Diagnosis and Counselling. Totowa, NJ: Humana Press, 2006, s. 852-856. ISBN 1-59259-956-7.
- Alessandro Castriota-Scanderbeg, Bruno Dallapiccola: Abnormal Skeletal Phenotypes: From Simple Signs to Complex Diagnoses. Springer, s. 849-850. ISBN 3-540-67997-9.
Linki zewnętrzne
- ROBERTS SYNDROME; RBS w bazie Online Mendelian Inheritance in Man (ang.)
- Roberts' pseudothalidomide syndrome w bazie Who Named It (ang.)