

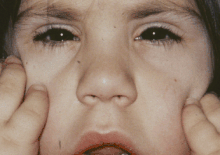
Zespół Weilla-Marchesaniego (ang. Weill-Marchesani syndrome, WMS) – rzadki, uwarunkowany genetycznie zespół wad wrodzonych, na który składają się niskorosłość, brachydaktylia, sztywność stawów i charakterystyczne wady narządu wzroku: mikrosferofakia (zbyt mała soczewka oka ma dodatkowo nieprawidłowy kształt), ektopia soczewki, ciężka krótkowzroczność i jaskra wrodzona. Dziedziczenie jest najczęściej autosomalne recesywne, rzadziej autosomalne dominujące.
Historia
Chorobę opisał jako pierwszy w 1932 roku francuski okulista Georges Weill u ośmiu swoich pacjentów. W 1939 roku kolejny opis przedstawił niemiecki okulista Oswald Marchesani. Przez pewien czas w użyciu była nazwa "zespołu Marchesaniego", ale przypomnienie pracy Weilla spowodowało, że do eponimu dodano też, w uznaniu pierwszeństwa, jego nazwisko. W 1955 Kloepfer i Rosenthal opisali dużą rodzinę z Luizjany, w której dodatkowo stwierdzili niskorosłość jako cechę heterozygotyczności względem allelu determinującego chorobę u homozygot. Wyjaśnienie natury genetycznego podłoża choroby doprowadziło do odkrycia, że zespół Weilla-Marchesaniego jest heterogennym etiologicznie zespołem, wywołanym przez mutacje w dwóch różnych genach: postać dziedziczona autosomalnie recesywnie przez mutacje w genie ADAMTS10, postać autosomalnie dominująco w genie FBN1. Mutacje w tym samym genie odpowiadają za fenotyp zespołu Marfana, zatem postać AD zespołu Weilla-Marchesaniego i zespół Marfana są schorzeniami allelicznymi.
Objawy i przebieg
Na obraz kliniczny zespołu składają się:
- brachydaktylia, nieprawidłowości śródręcza
- wady soczewki: mikrofakia, afakia, stożek soczewki
- zwichnięcie soczewki
- jaskra wrodzona
- zaćma
- miopatia
- niskorosłość lub karłowatość
- krótka szyja
- wady żeber, szeroka klatka piersiowa (puklerzowata)
- brachycefalia, płaska potylica, okrągła twarz
- wrodzone wady serca (stenoza zastawki płucnej, niedomykalność zastawki mitralnej, stenoza aortalna, przetrwały przewód tętniczy, ubytek przegrody międzykomorowej)
- utajony rozszczep kręgosłupa
- niekiedy łagodnego stopnia upośledzenie umysłowe.
Rozpoznanie
Możliwa jest diagnostyka prenatalna.
Etiologia
Postać dziedziczona autosomalnie recesywnie (częstsza) spowodowana jest mutacjami w obu allelach genu ADAMTS10 w locus 19p13.3-p13.2. Kodowane przez ten gen białko ADAMTS10 należy do proteaz macierzy pozakomórkowej (A Disintegrin and Metalloproteinase with Thrombospondin Motifs), a jego gen ulega ekspresji w skórze, płodowych chondrocytach i w tkance mięśnia sercowego (zarówno płodowej jak i dojrzałej). Badania mikroskopii elektronowej ujawniły nieprawidłowości w macierzy pozakomórkowej skóry u pacjentów z WMS. Postać dziedziczona autosomalnie dominująco związana jest z mutacją w jednym z alleli genu FBN1 w locus 15q21.1, kodującego fibrylinę-1. Część przypadków jest sporadyczna (niedziedziczona). Heterozygoty względem zmutowanego allela ADAMTS10 mogą mieć część nieprawidłowych cech zespołu, w tym niskorosłość i brachydaktylię.
Leczenie i rokowanie
Rokowanie jest dobre. Pacjenci z rozpoznanym zespołem Weilla-Marchesaniego powinni być objęci staranną opieką okulistyczną, w sztywności stawowej zalecana jest fizjoterapia.
Linki zewnętrzne
- WEILL-MARCHESANI SYNDROME, AUTOSOMAL RECESSIVE w bazie Online Mendelian Inheritance in Man (ang.)
- WEILL-MARCHESANI SYNDROME, AUTOSOMAL DOMINANT w bazie Online Mendelian Inheritance in Man (ang.)
- Weill-Marchesani syndrome w bazie Who Named It (ang.)