
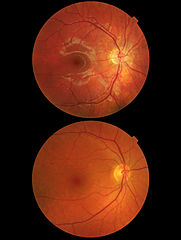
 Obraz dna oka, u góry obraz zwyrodnienia plamki żółtej, niżej prawidłowe dno oka | |
| Klasyfikacje | |
| ICD-10 | |
|---|---|


Zwyrodnienie plamki żółtej, zwyrodnienie plamki związane z wiekiem, dawniej: starcze zwyrodnienie plamki, AMD (od ang. age-related macular degeneration) – przewlekła, wieloczynnikowa, postępująca choroba oczu, występująca u osób po 50. roku życia. W wyniku tej choroby dochodzi do uszkodzenia siatkówki (a szczególnie jej części centralnej – plamki żółtej), co prowadzi do pogorszenia, ubytków, a niejednokrotnie całkowitej utraty widzenia centralnego, a w konsekwencji do ślepoty. Rozróżnia się postać suchą i wysiękową, która może dotknąć także ludzi młodych.
Jest jedną z najpowszechniejszych i nieodwracalnych przyczyn poważnej utraty widzenia centralnego, z praktyczną ślepotą włącznie. Szacuje się, że około 19% populacji europejskiej po 65. roku życia straci wzrok z powodu AMD. Rosnącą globalnie liczbę chorych tłumaczy się przede wszystkim wzrastającą średnią długością życia.
Objawy
Pierwsze oznaki są nieswoiste i trudne do uchwycenia przez chorego, szczególnie gdy dotyczą tylko jednego oka. Przez dłuższy czas schorzenie może rozwijać się powoli i bezobjawowo.
Do typowych objawów zwyrodnienia plamki żółtej należą: obniżenie ostrości widzenia (najczęściej zgłaszany wczesny objaw), widzenie prostych linii jako linii falistych lub zniekształconych, dyskomfort i trudności w czytaniu i pisaniu. Litery stają się przymglone, niewyraźne i zamazane, a przedmioty zniekształcone (metamorfopsja). Objawem późnej fazy choroby jest mroczek centralny: zaciemnienie środka pola widzenia. Postęp powoduje stopniowe zwiększanie się mroczka, czemu towarzyszy zanik ostrości, kontrastu i barw w pozostałej części pola widzenia. Choroba postępuje w różnym tempie i może prowadzić nawet do całkowitej utraty wzroku.
Choroba rozwija się zazwyczaj obuocznie, ale niekoniecznie w tym samym tempie i nasileniu. Według wielu autorów w ciągu roku od rozpoznania wysiękowego AMD w jednym oku istnieje 5–15% ryzyko rozwinięcia się zmian w drugiej gałce.
Czas rozwoju choroby wynosi od kilku tygodni do kilku lat. Liczba pacjentów niewidomych z powodu AMD osiąga szczyt między 70. a 84. rokiem życia.
Wieloletnie obserwacje pacjentów z AMD wskazują na częściej występujące inne schorzenia, jak udar mózgu czy krwotok śródmózgowy. Zaawansowane AMD zwiększa też ryzyko wystąpienia choroby niedokrwiennej serca.
Jedna trzecia chorych cierpi na zaburzenia emocjonalne związane z utratą samodzielności. Pogorszenie jakości życia pacjentów z zaawansowanym AMD wynika z głębokiego spadku widzenia osiowego (niemożność czytania, pisania, rozpoznawania szczegółów twarzy). Widzenie obwodowa pozostaje nienaruszone, więc osoby dotknięte AMD nie wymagają szczególnej opieki w poruszaniu się poza domem. U ponad 60% pacjentów z obustronnym AMD występuje zwiększenie ryzyka urazów i ich powikłań.
Etiopatogeneza
Choroba polega na tworzeniu się nowych patologicznych naczyń krwionośnych oraz ucieczkę składników morfotycznych i białek poza już istniejące naczynia. Oba procesy powodują proces zapalny i powstawanie druz.
Obraz choroby obejmuje zmiany w fotoreceptorach, nabłonku barwnikowym siatkówki (RPE), błonie Brucha i warstwach choriokapilar. Sednem objawów i skutków choroby jest jednak postępująca degeneracja i atrofia komórek nabłonka barwnikowego, co prowadzi w końcu do nieodwracalnych uszkodzeń fotoreceptorów.
Przyczyny powstawania zwyrodnienia plamki żółtej są słabo poznane. Są wieloczynnikowe i obejmują złożone procesy metaboliczne, funkcjonalne, genetyczne i środowiskowe.
W przebiegu i rozwoju choroby można wyróżnić cztery procesy:
- lipofuscynogenezę - powstawanie lipofuscyny w komórkach RPE;
- Naukowcy są zgodni, że uszkodzenia komórek RPE jest wczesnym i głównym etapem rozwoju zwyrodnienia. Uzasadnia się to tym, że RPE pełni kluczową rolę w funkcjach metabolicznych fotoreceptorów (utrzymywanie bariery krew-siatkówka, udział w cyklu wzrokowym, fagocytoza złuszczonych POS - końcowych fragmentów fotoreceptorów). Wraz z wiekiem komórki RPE, szczególnie postmiotyczne (nieodnawialne), stają niewydolne fagocytarno-metabolicznie. Powoduje to, że w komórce stopniowo odkłada się lipofuscyna. Jej składnikami są lipidy i białka pochodzące z fagosomów, lizosomów i POS. Ulegają one procesom oksydacji, która w komórkach oka ma sprzyjające warunki - wysokie stężenie tlenu i ekspozycja na promieniowanie UVA. Jednym ze znanych produktów jest A2E - bis-retinoid pirymidynowy, o udowodnionej fotocytotoksyczności. Jest on silnym generatorem wolnych rodników.
- druzogenezę - powstawanie nierozpuszczalnych druz;
- Druzy są bezpostaciowymi zewnętrzkomórkowymi złogami lipidowo-białkowymi. Odkładają się między RPE a wewnętrzną (kolagenową) warstwą błony Brucha. Wyróżnia się druzy miękkie i twarde. Nieliczne małe (< 63 μm) twarde druzy znajdowane są u 95% ludzi starszych. Za główny czynnik zaawansowanej choroby uważa się liczne większe (> 125 μm), a szczególni miękkie zlewające się (>250 μm) druzy. Ryzyko jest większe, jeśli druzom, nawet małym, towarzyszą zmiany barwnikowe. Wykazano, że zmiany degeneracyjne związane są obecnością druz w pobliżu zdegenerowanych komórek RPE. Proces ich powstawania jest złożony i wieloczynnikowy. W badaniu oftalmoskopowym nie ma możliwości zobaczenia ich na wczesnym etapie tworzenia (rozmiar poniżej 30 μm).
- Druzy wpływają negatywnie na komórki RPE dwojako. Po pierwsze, stanowią fizyczną przeszkodę między komórkami RPE a choriokapilarami. Wywołują odwarstwienie się zależnych od siebie struktur i upośledzają wymianę składników pokarmowych i produktów przemiany materii. Po drugie, inicjują lokalny, przewlekły proces zapalny i uruchamiają działanie układu immunologicznego.
- Wśród zidentyfikowanych białkowych składników druz są pozostałości komórek RPE, komponenty układu odpornościowego (np. wypustki komórek dendrytycznych, cząsteczki zgodności tkankowej, aktywatory i inhibitory układu dopełniacza) oraz tak zwany kompleks atakujący błonę. Kompleks ten powoduje unicestwienie obcych antygenów, ale w długim czasie także własnych tkanek - komórek RPE, fotoreceptorów i innych tkanek oka. Jest wysoce prawdopodobne, że lokalny proces zapalny, aktywacja układu dopełniacza i powstawanie kompleksu atakującego błonę są kluczowymi czynnikami druzogenezy, a następnie fragmentacji błony Brucha, co w końcu może wywoływać neowaskularyzację.
- zapalenie
- neowaskularyzację naczyniówkową (choroidal neovascularization, CNV) - nowotworzenie naczyń krwionośnych.
- Neowaskularyzacja naczyniówkowa (podsiatkówkowa) jest odpowiedzialna za powstawanie wyniesień i zgrubień w siatkówce. Są one widoczne na dnie oka w badaniu oftalmoskopowym. Nowo utworzone naczynia rozprzestrzeniają się od naczyń włosowatych naczyniówki, przez ubytki w błonie Brucha, do przestrzeni pod nabłonkiem barwnikowym RPE, aż do warstwy neurosensorycznej siatkówki. Patologiczne naczynia są wyraźnie kręte, słabe i kruche. Z tego powodu często są nieszczelne - powstają liczne wynaczynienia krwi. Wysięki te, wraz ze skrzepami i bliznami, powodują następnie zaburzenia widzenia i ubytki widzenia centralnego. CNV może też poprzedzać lub następować po odwarstwieniu RPE.
- W prawidłowych warunkach komórki śródbłonka (endotelialne) wyściełające światło naczyń krwionośnych znajdują się w stanie dynamicznej równowagi utrzymywanej przez czynniki proangiogenne (VEGF, FGF, PDGF) i antyangiogenne (PEDF, angiostatyna, trombospondyna, endostatyna). Czynniki te precyzyjnie kontrolują unaczynienie. Zaburzenie tej równowagi, przez wzmocnienie czynników angiogennych lub osłabienie czynników antyangiogennych, powoduje zainicjowanie neowaskularyzacji. W przypadku AMD czynnikiem zaburzającym wydaje się być lokalny proces zapalny z towarzyszącymi mu reakcjami immunologicznymi. Obecne przy stanie zapalnym neutrofile, makrofagi i komórki tuczne są znane ze zdolności wytwarzania i uwalniania wielu czynników proangiogennych. Najistotniejszą rolę dla CNV wydają się mieć VEGF i PEDF.
Postacie choroby
Uwzględniając cechy kliniczne i patofizjologiczne wyróżnia się dwie postaci zwyrodnienia plamki żółtej:
- sucha (zanikowa; zaniku geograficznego i/lub druz; niewysiękowa)
- mokra (wysiękowa; postać podsiatkówkowej błony neowaskularnej; neowaskularna)
Postać sucha
Stanowi około 85-90% przypadków. Postać sucha, łagodniejsza, polega na stopniowej degeneracji komórek nabłonka barwnikowego siatkówki (RPE), na skutek powstawania coraz liczniejszych i zlewających się druz - złogów między RPE a błoną Brucha. W czasie postępu choroby dochodzi do miejscowych zaników komórek fotoreceptorowych (zanik geograficzny), a w końcu do zaniku komórek nabłonka barwnikowego.
Zanikiem geograficznym określa się wyraźnie odgraniczony, okrągły lub owalny obszar hipo- lub depigmentacji siatkówki w plamce, o średnicy co najmniej 175 μm. Spowodowany jest zanikiem w warstwie RPE fotoreceptorów i naczyń włosowatych naczyniówki (choriokapilar). Zmiany te powodują powolny spadek ostrości wzroku, upośledzenie widzenia przy gorszym oświetleniu, niemożność utrzymania fiksacji i ograniczenie obszaru dobrego widzenia, co uniemożliwia przeczytanie rzędu liter w tekście.
Badanie okulistyczne nie musi wykazywać spadku ostrości, gdyż wyspowy charakter zmian pozwala pacjentowi nadal krótkotrwale fiksować obraz. Objawia się stopniowym, stosunkowo wolnym, pogarszaniem widzenia o małym lub średnim nasileniu przez okres miesięcy lub lat.
Postać mokra
Znacznie poważniejsza jest forma mokra AMD. Występuje ona u około 10% chorych, ale odpowiada za prawie 90% przypadków poważnej utraty wzroku. Chory doświadcza podobnych objawów chorobowych, ale są one bardziej nasilone i szybciej postępujące. W jej przebiegu dochodzi do nieprawidłowej angiogenezy (neowaskularyzacji) - wytwarzane są dodatkowe, nieprawidłowe naczynia krwionośne, które przerastają siatkówkę i uszkadzają komórki. Nowe naczynia są poskręcane, słabe i nieszczelne, co powoduje wysięki i krwotoki. Może ona pojawiać się jako postać izolowana (tylko cechy postaci mokrej), ale u około ⅓ wszystkich pacjentów z neowaskularyzacją stwierdza się również obecność druz, przegrupowań barwnika i zaniku nabłonka barwnikowego.
Przebiega ona w sekwencji trzech zdarzeń:
- neowaskularyzacja podsiatkówkowa (naczyniówkowa) - jej źródłem są naczynia warstwy choriokapilarnej
- surowicze lub włóknisto-naczyniowe odwarstwienie nabłonka barwnikowego siatkówki
- powstanie tarczowatej blizny
Postać niewysiękowa może przejść w postać wysiękową, czemu towarzyszy gwałtowne nasilenie się objawów. Progresji sprzyja palenie tytoniu i niewyrównane ciśnienie tętnicze
Nieleczona postać wysiękowa w ciągu średnio 2 lat (z dużym zróżnicowaniem osobniczym: od 6 miesięcy do 10 lat) doprowadza do powstania blizny tarczowatej wskutek włóknienia tkanek w obrębie CNV.
Czynniki ryzyka
Do najważniejszych czynników ryzyka należą:
- wiek pacjenta: powyżej 50. roku życia
- Choroba dotyczy 5–10% osób w wieku 65–75 lat i 20–30% osób w wieku ponad 75 lat (współczynniki te odnoszą się do krajów wysoko rozwiniętych i wahają się w zależności od badania i przyjętej definicji choroby)
- palenie tytoniu (nawet w przeszłości) - zwiększa ryzyko 2,5-krotnie
- inne choroby układu sercowo-naczyniowego (np. nadciśnienie tętnicze)
- płeć - niektóre badania sugerują większą podatność kobiet
- rasa biała,
- czynniki genetyczne - występowanie AMD w rodzinie
- niebieska tęczówka
- dalekowzroczność.
Na wpływ na rozwój choroby może mieć też wieloletnie narażenie na intensywne światło (np. podczas pracy na świeżym powietrzu) oraz dieta (niedobór przeciwutleniaczy, np. witaminy C, witaminy E, beta-karotenu, selenu).
Rola czynników genetycznych
Wpływ czynników genetycznych na rozwój AMD jest bardzo prawdopodobny. Wiele badań wskazuje na rodzinne występowanie choroby. Jednak na razie nie udało się zidentyfikować pojedynczych genów mających kluczowy wpływ na zapadalność na AMD. Zidentyfikowano wiele genów (np. ABCA4, ELOVL4, FIBL-6, SOD2, APOE), których mutacje powodują objawy zbieżne z obrazem klinicznym AMD, jednak nie są w pełni odpowiedzialne za etiopatogenezę choroby.
Wykazano że, mutacje w genach kodującym wchodzące w skład układu dopełniacza czynnik H i czynnik B mogą w znaczny sposób zwiększać ryzyko zachorowania na AMD.
Kilka niezależnych grup badawczy zidentyfikowało wspólny wariant genu CFH czynnika H i B układu dopełniacza. Występowanie tej odmiany można wykazać u około 50% pacjentów z pełnymi objawami choroby. Mutacja czynnika H polega na zamianie aminokwasu tyrozyny na histydynę w łańcuchu polipeptydowym w pozycji 402 (Y402H).Mutacja ta hamuje reakcję zapalną, która powstaje w wyniku działania składnika C3b (i alternatywnej drogi aktywacji dopełniacza), zarówno będąc kofaktorem nieuczynnienia C3b do formy nieaktywnej C3bi, a także przez osłabienie siły wiązania się C3b i czynnika B w kompleks. W warunkach fizjologicznych białko C reaktywne (CRP) oraz polianionowe cząsteczki powierzchniowe takie jak glikozoaminoglikany zwiększają hamujący wpływ czynnika H na dopełniacz. Lecz mutacja w genie CFH (Tyr402His) powoduje zmniejszenie powinowactwa czynnika H do CRP i prawdopodobnie też zmienia jego zdolność rozpoznawania glikozoaminoglikanów. Te czynniki powodują zmniejszenie regulacyjnej czynności czynnika H na dopełniacz na dnie oka co przyczynia się do powstawania patologicznej reakcji zapalnej w plamce żółtej, i co najpewniej wywołuje druzogenezę.
Postać suchą powiązano ze zmniejszoną aktywnością enzymu DICER1 w siatkówce. W badaniach in vivo na myszach ustalono, że odpowiada on za niszczenie Alu RNA - małych fragmentów materiału genetycznego. Brak enzymu powodował zmiany w siatkówce odpowiadające suchej postaci zwyrodnienia plamki żółtej.
Opublikowane w 2006 r. badania wykryły znaczenie przyczynowe mutacji w genie HTRA1 dla rozwoju AMD.
Częstość występowania
Zdiagnozowanych chorych na różne postacie AMD jest na świecie co najmniej 20-30 milionów. Szacunkowa liczba osób niezdiagnozowanych to ok. 40 milionów. Na świecie notuje się około 0,5 mln nowych przypadków rocznie. Statystyki pokazują wzrastającą zapadalność na AMD.
Degeneracja plamki żółtej jest główną przyczyną utraty wzroku u ludzi powyżej 50. roku życia. Występuje częściej u kobiet. Na całym świecie liczba chorych sięga 50 mln. W związku ze starzeniem się społeczeństwa problem AMD narasta, a schorzeniem zagrożonych jest ok. 10% osób po 45. roku życia. Z tego względu bywa określana chorobą cywilizacyjną XXI wieku.
Diagnostyka
U pacjenta z podejrzeniem AMD wykonuje się pełne badanie okulistyczne: badanie ostrości wzroku do dali i bliży (tablice Snellena i specjalne tablice ETDRS) oraz wziernikowanie dna oczu przy poszerzonych źrenicach. Dodatkowo stosuje się jeszcze test poczucia kontrastu Pelli-Robsona, pomiar ciśnienia śródgałkowego, test Amslera. Zaletą tego ostatniego jest to, że pacjenci mogą wykonać go samodzielnie, nawet w warunkach domowych.
Diagnostyka rozszerzana jest o nieinwazyjną tomografię oka, umożliwiającą dokładne pomiary wielkości poszczególnych patologii. Druzy mogą być widoczne w badaniu jako hiperrefleksyjne uniesienia w warstwie RPE. Uwapnione druzy mogą powodować cieniowanie. Zanik geograficzny jest widoczny jako ubytek warstwy siatkówki (wzmocnienie odbić z naczyniówki). OTC służy też do monitorowania postępów choroby i wskazywania ponownej iniekcji leków.
Zobrazowanie naczyń krwionośnych realizowane jest metodami angiografii fluoresceinowej (AF) lub indocyjaninowej (ICG) – badania służące zobrazowaniu naczyń krwionośnych. ICG stosuje się przeważnie w celu obrazowania głębszych warstw naczyniówki, szczególnie przy nieklasycznych postaciach mokrej AMD.
W obrazie angiografii neowaskularyzacja postaci mokrej AMD obserwowana jest w 3 rodzajach:
- klasycznej - obraz przypomina koronkę; plama jest dobrze widoczna od początku badania, a przeciek fluoresceiny następuje później. Daje to obraz jasnej plamy, nie mającej ostrych granic.
- włóknisto-naczyniowe odwarstwienie RPE - obserwuje się wolnonarastającą hiperfluorescencję występującą 30-60 sekund po podaniu barwnika do żyły łokciowej. Hiperfluorescencja nasila się i jest trudna do prześledzenia.
- późny przeciek o nieustalonym źródle - wolnonarastająca hiperfluorescencja między 2 a 5 minutą badania.
W obu postaciach choroby w badaniu dna oka obserwowane są druzy i zaburzenia pigmentacji siatkówki (hipo- i hiperpigmentacja).
AMD bywa źle rozpoznawane jako retinopatia cukrzycowa lub inne schorzenia styku ciało szkliste - siatkówka.
Monitorowanie przebiegu AMD jest także możliwe przy użyciu perymetrów (polomierzy) komputerowych wykonanych w technologii PHP i wykorzystujących zjawisko Verniera.
Dodatkowo istnieją aplikacje na smartfony zawierające testy i badania pozwalające wstępnie zdiagnozować oraz monitorować schorzenie. W kwietniu 2015 roku amerykańska Agencja Żywności i Leków zaaprobowała aplikację mobilną do monitorowania AMD, która pozwala pacjentowi wykonać krótkie, kilkuminutowe badania kontrolne w domu (częstotliwość badań określa lekarz, zazwyczaj dwa razy w tygodniu). Aplikacja automatycznie przesyła wyniki do specjalnego portalu, do którego poufny dostęp ma okulista prowadzący. W razie znaczącej zmiany w wynikach badania, lekarz otrzymuje alert (powiadomienie). Aplikacja bada czynniki, które mają wpływ nie tylko na przebieg AMD, ale także retinopatii cukrzycowej.
Leczenie
Działania prewencyjne i zapobiegawcze obejmują przede wszystkim prozdrowotne zalecenia dotyczące trybu życia (zmniejszenie masy ciała, zmniejszenie spożycia alkoholu, ograniczenie tłuszczów zwierzęcych, zwiększenie podaży nienasyconych kwasów tłuszczowych; dieta śródziemnomorska). Kluczowym modyfikowalnym czynnikiem ryzyka jest palenie tytoniu. Należy również wzbogacić dietę w antyoksydanty i minerały: witamina C, witamina E, cynk, glutation, a przede wszystkim luteina i zeaksantyna. Wykazano, że te luteina i zeaksantyna (związki karotenoidowe obecne w fotoreceptorach i komórkach RPE) poprawiają jakość widzenia. Chronią również plamkę żółtą przed wpływem stresu oksydacyjnego, są bowiem zdolne do unieczynnienia tlenu singletowego. Blokują również fotooksydację lipofuscyny. Organizm człowieka nie wytwarza tych dwóch związków i muszą być one dostarczane z pokarmem. Dalszych badań wymaga precyzyjne ustalenie optymalnego składu mieszanek suplementacyjnych i dawek każdego ze związków.
W ramach amerykańskich badań AREDS (Age-Related Eye Disease Study) osiągnięto 25% ograniczenie ryzyka progresji AMD do postaci zaawansowanej poprzez podawanie wysokich dawek przeciwutleniaczy (witaminy C, E, beta-karotenu), tlenku cynku i tlenku miedzi. Zestaw podawanych substancji zależał od genów CFH (skutecznie było podawanie tylko przeciwutleniaczy) i ARMS2 (skuteczne było podawanie tylko tlenków), które powiązano ze zwiększonym ryzykiem zaawansowanej formy schorzenia.
Obiecującą metodą leczenia postaci suchej jest również przeszczepienie komórek nabłonka barwnikowego w przestrzeń podsiatkówkową. Komórki takie uzyskuje się z różnych typów komórek macierzystych. Metoda ma jednak nadal wiele wad uniemożliwiających zastosowanie kliniczne. Wymaga również podawania leków immunosupresyjnych po transplantacji.
Jedną z wczesnych metod leczenia była terapia fotodynamiczna (Photodynamic Therapy, PDT). Polega ona na podaniu substancji fotouczulającej, werteporfiryny, wiążącej się z lipoproteinami o niskiej gęstości, które zaś łatwo wiążą się z nowo tworzonymi naczyniami krwionośnymi - źródłem degeneracji siatkówki w wysiękowej postaci AMD. Miejsca gromadzenia się werteporfiryny naświetlane są następnie laserem o długości fali 689 nm. Aktywacja substancji prowadzi do powstania wysoko reaktywnych form tlenu (tlen singletowy), prowadzących do powstania zakrzepów i zamknięcia niepożądanych naczyń. Metoda jest szeroko stosowana z dobrymi skutkami. W badaniach klinicznych stwierdzono istotne pogorszenie ostrości wzroku po 18 miesiącach u 60% nieleczonych, a tylko u 25% pacjentów leczonych PDT. Jest to jednak terapia objawową i ma wiele efektów ubocznych, w tym utratę widzenia (4% przypadków). Co więcej, wskazania do PDT są rygorystyczne. Wielu pacjentów z postacią wysiękową nie może z niej skorzystać. Niektóre badania pokazują, że PDT może powodować wzrost ekspresji VEGF - silnego aktywatora neowaskularyzacji.
Inną metodą z zastosowaniem lasera jest koagulacja termiczna (thermal laser photocoagulation, TLP). Laseru używa się tam do niszczenia ognisk patologicznych naczyń krwionośnych. Jej stosowanie jest jednak ograniczone wyłącznie do ognisk pozadołkowym.
Termoterapia przezźrenicza (transpupillary thermotherapy, TTT) wykorzystuje laser diodowy o długości fali 810 nm do zamykania światła niepożądanych naczyń. Wiązka obejmuje ognisko neowaskularyzacji i wywołuje w nim wzrost temperatury o 5-10 °C, skutkujący uszkodzeniem śródbłonka naczyń, zakrzepami przyściennymi i w końcu zamknięciem światłą naczynia.
Inną metodą są kuracje anty-VEGF. VEGF, to czynnik wzrostu śródbłonka naczyniowego stymulujący neowaskularyzację naczyniówkową (choroidal neovascularisation, CNV). Terapia opiera się na miejscowym podawaniu substancji blokujących działanie VEGF. Stosuje się w postaci iniekcji doszklistkowych lub okołotwardówkowych z dobrym skutkiem następujące leki:
- pegaptanib (Macugen) - aptamer mający duże powinowactwo z główną izoformą VEGF. Wiele randomizowanych badań z podwójnie ślepą próbą jednoznacznie wykazują, że ma on statystycznie istotny, korzystny efekt terapeutyczny w leczeniu AMD. Pod koniec 2004 r. został zatwierdzony przez FDA jako lek spowalniający utratę widzenia we wszystkich podtypach mokrej AMD, z zaleceniem stosowania dawki 0,3 mg w iniekcji do ciała szklistego, co 6 tygodni.
- ranibizumab (Lucentis) - fragment przeciwciała anty-VEGF blokujący wszystkie izomery VEGF. Podawany w iniekcji do ciała szklistego. Próby kliniczne dowiodły skuteczności poprzez hamowanie wysięgów i poprawę ostrości widzenia. W 2006 r. zatwierdzony w USA do terapii wysiękowego AMD.
- bewacyzumab (Avastin) - przeciwciało monoklonalne wiążące się ze wszystkimi izoformami VEGF-A. Nie został on zatwierdzony do jako lek w chorobach oczu, ale jest stosowany w wysiękowej postaci AMD na zasadzie off-lable, jako że jest dostępny na rynku farmaceutyczny. Próby kliniczne z dawkami 1 mg i 1,25 mg w iniekcjach do ciała szklistego wykazały poprawę ostrości widzenia i zmniejszenie wysięków u większości chorych.
- triamcynolon (Polcoltoron) - dostępny na rynku steroid o działaniu przeciwzapalnym. Wykazuje korzystne działanie w wielu zaburzeniach wewnętrzgałkowych, w tym obrzękach i procesach rozrostowych.
- aflibercept (Eylea, Zaltrap) - rekombinowane białko fuzyjne wiążące się z naczyniowo-śródbłonkowym czynnikiem wzrostu A (VEGF-A); związek typu VEGF-trap.
W terapii neowaskularnej AMD mogą znaleźć zastosowanie również endogenner czynniki antygiogenne, jak angiostatyna, endostatyna i PEDF. W 2008 peptyd AdPEDF, typu PEDF, był w trakcie I fazy badań klicznicznych.
Do leczenia AMD przeznaczone były również: octan anekortawu, skwalamina, Sirna-027 (wstrzymany w II fazie badań klinicznych). Nie przeszły one jednak wszystkich niezbędnych do rejestracji prób klinicznych.