
| microcephalia | |
 dziecko z mikrocefalią (po lewej) w porównaniu do dziecka z prawidłową wielkością głowy | |
| Klasyfikacje | |
| ICD-10 | |
|---|---|
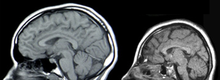
Mikrocefalia (małogłowie, łac. microcephalia, ang. microcephaly) – wada rozwojowa charakteryzująca się nienaturalnie małymi wymiarami czaszki (a więc również puszki mózgowej). Małogłowie rozpoznaje się gdy obwód głowy, mierzony pomiędzy okolicą tuż nad wałami nadoczodołowymi do najbardziej ku tyłowi wysuniętej części kości potylicznej (opisthocranion), nie przekracza wartości średniej dla płci i wieku w danej populacji pomniejszonej o 3 odchylenia standardowe (SD). Mikrocefale mają masę mózgu mniejszą niż 900 g. Mikrocefalia może wystąpić jako wada izolowana lub element zespołu wad.
Etiologia
Przyczyny małogłowia dzielimy na genetyczne i niegenetyczne. Najczęstsze niegenetyczne przyczyny mikrocefalii to:
1. Nieinfekcyjne choroby ośrodkowego układu nerwowego (OUN) w okresie prenatalnym i perinatalnym
- Niedokrwienie OUN
- Krwawienie do OUN
- Urazy OUN
2. Zakażenia wewnątrzmaciczne
3. Działanie związków chemicznych i leków
- płodowy zespół alkoholowy
- palenie tytoniu przez matkę
- zażywanie przez matkę leków przeciwpadaczkowych (hydantoiny, karbamazepiny, fenobarbitalu)
4. Choroby matki w ciąży
5. Inne czynniki
- niedożywienie matki.
Najważniejsze genetyczne zespoły, w których obrazie klinicznym występuje mikrocefalia:
- Zespół Pataua – trisomia 13
- Zespół Edwardsa – trisomia 18
- Zespół Downa – trisomia 21
- delecja 5p (zespół cri du chat)
- delecja 4p (zespół Wolfa-Hirschhorna)
- zespół de Grouchy’ego
- zespół Millera i Diekera (OMIM 247200)
- zespół Angelmana
- zespół Williamsa
- holoprozencefalia
- zespół Smitha-Lemliego-Opitza
- lissencefalia
- zespół Seckela
- zespół Blooma
- zespół Nijmegen
- zespół Retta
- zespół Rubinsteina-Taybiego
- rodzinna postać małogłowia dziedziczona autosomalnie dominująco (OMIM 156580)
- rodzinna postać małogłowia dziedziczona autosomalnie recesywnie, typy 1-6
- choroby metaboliczne, np. fenyloketonuria.
- wczesna dziecięca encefalopatia padaczkowa typu 13 (EIEE13) powiązana z patogenną mutacja w genie SCN8A
Zobacz też
Bibliografia
- Lech Korniszewski: Dziecko z zespołem wad wrodzonych. Diagnostyka dysmorfologiczna. Wyd. II, PZWL 2005 ISBN 83-200-3042-0.