
Serpiny – nadrodzina białek o podobnej strukturze, zidentyfikowanych po raz pierwszy dzięki ich aktywności hamowania proteaz. Występują we wszystkich królestwach organizmów żywych. Akronim stanowiący ich nazwę ukuto, ponieważ pierwsze zidentyfikowane serpiny działały jako inhibitory proteaz serynowych przypominających chymotrypsynę (serine protease inhibitors). Znane są z niezwykłego mechanizmu działania, inhibicji nieodwracalnej. Doprowadzają proteazy do znacznej zmiany konformacji, niszcząc miejsce aktywne. Kontrastuje to z bardziej powszechnym mechanizmem inhibicji kompetycyjnej proteaz, w którym inhibitor przyłącza się do miejsca aktywnego enzymu i blokuje je.
Hamowanie proteaz przez serpiny kontroluje szereg procesów biologicznych, jak krzepnięcie czy zapalenie, w efekcie czego białka te stanowią przedmiot badań medycznych. Ich unikalna zmiana konformacyjna czyni je także przedmiotem zainteresowania biologii strukturalnej i badań zwijania białek. Mechanizm zmiany konformacyjnej daje pewne zalety, ale również i wady: geny serpin są podatne na mutacje, mogące skutkować serpinopatiami, jak niewłaściwe fałdowanie się białka czy tworzenie się nieaktywnych długołańcuchowych polimerów. Polimeryzacja serpin nie tylko ogranicza ilość dostępnego aktywnego inhibitora, ale również prowadzi do odkładania się polimerów, co skutkuje śmiercią komórki i dysfunkcją narządu.
Choć większość serpin kontroluje kaskady proteolityczne, niektóre z białek o budowie serpiny nie są inhibitorami enzymów, pełniąc zamiast tego różne funkcje, jak magazynowanie białka (jak w przypadku owoalbuminy białka jaja), transport (jak białka transportujące hormony, np. TBG, transkortyna) czy opiekowanie się innymi białkami (jak HSP47). Terminem serpin określa się je wszystkie pomimo odmiennej funkcji, ponieważ wiąże je pokrewieństwo ewolucyjne.
Historia
Aktywność inhibitora proteaz w osoczu krwi po raz pierwszy odnotowano pod koniec pierwszej dekady XIX wieku, ale izolacja serpin doszła do skutku dopiero w latach 50. XX wieku – były to antytrombina i alfa1-antytrypsyna. Początkowe badania skupiały się na ich roli w chorobach człowieka. Niedobór alfa1-antytrypsyny należy do najczęstszych chorób genetycznych, powoduje rozedmę płuc. Niedobór antytrombiny powoduje zakrzepicę.
W latach 80. XX wieku stało się jasne, że inhibitory te stanowią część nadrodziny spokrewnionych ze sobą białek, do której zaliczają zarówno inhibitory proteaz (alfa1-antytrypsyna) i białka bez takiej aktywności (owoalbumina). Termin serpina (serpin) utworzono, bazując na najczęstszej w obrębie tej nadrodziny aktywności inhibitorów proteaz serynowych (serine protease inhibitors). Mniej więcej w tym samym czasie odkryto strukturę pierwszych serpin, wpierw w konformacji zrelaksowanej, potem w naprężonej. Struktura wskazywała, że mechanizm inhibicji obejmuje niezwykłą zmianę konformacyjną, co zwróciło uwagę na badania strukturalne serpin.
Dotychczas zidentyfikowano powyżej 1000 serpin, w tym 36 białek ludzkich. Serpiny występują we wszystkich królestwach organizmów żywych: w organizmach zwierząt, grzybów, roślin, bakterii i archeonów, a nawet w pewnych wirusach (pokswirusy). W pierwszej dekadzie XXI wieku wprowadzono systematyczne nazewnictwo w celu kategoryzacji członków nadrodziny w oparciu o pokrewieństwo ewolucyjne. Serpiny stanowią największą i najbardziej zróżnicowaną nadrodzinę inhibitorów proteaz.
Aktywność

Większość serpin to inhibitory proteaz, których target molekularny stanowią zewnątrzkomórkowe proteazy serynowe typu chymotrypsyny, a więc enzymy posiadające w centrum aktywnym triadę katalityczną obejmującą nukleofilową resztę seryny. Przykłady obejmują trombinę, trypsynę i ludzką elastazę neutrofilową. Serpiny prowadzą inhibicję nieodwracalną, także dla cząsteczki inhibitora, poprzez uwięzienia półproduktu mechanizmu katalitycznego.
Pewne serpiny hamują inne rodziny proteaz, zazwyczaj w takim wypadku proteazy cysteinowe, określa się je terminem „cross-class inhibitors” (inhibitorów międzyklasowych). Enzymy te różnią się od proteaz serynowych wykorzystywaniem w miejscu aktywnym nukleofilowej reszty cysteinylowej zamiast serylu. Niemniej chemia reakcji enzymatycznej jest podobna, a mechanizm inhibicji obu klas enzymów przez serpiny taki sam. Przykłady takich serpin to SERPINB4, antygen 1 rak kolczystokomórkowego skóry (SCCA-1) i ptasie białko MENT (myeloid and erythroid nuclear termination stage-specific protein), hamujące proteazy cysteinowe typu papainy.
Funkcja biologiczna i lokalizacja
Inhibicja proteaz
Około dwóch trzecich ludzkich serpin działa pozakomórkowo, hamując proteazy w krwiobiegu, bądź modulując ich aktywności. Pozakomórkowe serpiny na przykład regulują proteolityczne kaskady kluczowe dla procesu krzepnięcia krwi (antytrombina) czy zapalenia i odpowiedzi immunologicznej (antytrypsina, alfa1-antychymotrypsyna, inhibitor C1) remodelingu tkankowego (PAI-1). Poprzez inhibicję proteaz kaskad sygnałowych mogą również wpływać na procesy rozwojowe.
Trudno jest zidentyfikować proteazy stanowiące wewnątrzkomórkowe targety molekularne serpin, jako że wiele z tych cząsteczek wydaje się pełnić przenikające się role. Co więcej, wiele ludzkich serpin nie posiada dokładnych funkcjonalnych odpowiedników w organizmach modelowych, jak mysz. Niemniej ważną funkcją wewnątrzkomórkowych serpin może być ochrona przed nieprawidłową aktywnością proteaz w komórce. Na przykład jedna z najlepiej opisanych ludzkich wewnątrzkomórkowych serpin SERPINB9 (serpina B9) hamuje proteazę cytotoksycznych ziarnistości granzym B. Poprzez to serpina B9 może ochraniać komórkę przed niecelowym uwolnieniem granzymu B i przedwczesną bądź niepożądaną apoptozą.
Niektóre wirusy wykorzystują serpiny do wypaczania działania proteaz ich gospodarzy. Serpina wirusa ospy krowiej CrmA (cytokine response modifier A) służy mu do unikania reakcji zapalnej i apoptozy w zainfekowanych komórkach gospodarza. CrmA wzmaga infekcyjność poprzez supresję odpowiedzi zapalnej gospodarza przez hamowanie działania interleukiny 1 i 18 umożliwianego przez będącą proteazą kaspazę 1. U eukariotów roślinna serpina hamuje zarówno metakaspazy, jak i proteazy cystynowe typu papainy.
Inne funkcje
Niebędące inhibitorami serpiny wewnątrzkomórkowe wykonują szeroki zakres ważnych zadań. TBG (globulina wiążąca tyroksynę) i transkortyna transportują hormony odpowiednio tyroksynę i kortyzol. Owoalbumina to najobficiej występujące białko białka jaj. Jego dokładna funkcja nie jest znana. Uważa się, że stanowi materiał zapasowy dla rozwijającego się zarodka. Białko szoku cieplnego HSP 47 jest szaperonem, kluczowym dla fałdowania się kolagenu. Stabilizuje ono potrójną helisę kolagenu podczas obróbki w siateczce śródplazmatycznej.
Pewne serpiny zarówno stanowią proteazy, jak i odgrywają dodatkowe role. Przykładowo inhibitor jądrowej proteazy cystynowej MENT u ptaków uczestniczy także w remodelingu chromatyny w ptasich erytrocytach.
Struktura

Wszystkie serpiny łączy wspólna budowa, choć wypełniają różne funkcje. Wszystkie zazwyczaj mają 3 harmonijki β (oznaczane literami A, B i C) oraz 8 lub 9 helis α (oznaczanych hA–hI). Najistotniejsze dla funkcji serpin są harmonijka A oraz pętla obejmująca centrum aktywne (reactive centre loop, RCL). Harmonijka A obejmuje 2 pasma β ułożone równolegle z regionem określanym jako shutter oraz wyższym regionem nazywanym breach. RCL rozpoczyna interakcję z docelową proteazą. RCL może być w pełni wystawione bądź częściowo schowane w harmonijce A. Serpina znajduje się w stanie dynamicznej równowagi pomiędzy tymi dwoma stanami. RCL reaguje z resztą struktury tylko czasowo, wobec czego zachowuje znaczną giętkość, wyeksponowane na działanie rozpuszczalnika.
Struktura serpin obejmuje kilka różnych konformacji, których ustalenie jest konieczne dla zrozumienia ich wielostopniowego mechanizmu działania. Biologia strukturalna gra wobec tego kluczową rolę w zrozumieniu mechanizmu i biologii serpin.
Zmiana konformacyjna i mechanizm inhibicji
Stanowiące inhibitory serpiny nie hamują swych targetów poprzez typowy mechanizm kompetycyjny wykorzystywany przez większość niewielkich inhibitorów proteaz (inhibitor typu Kunitz). Zamiast tego serpiny wykorzystują zmianę konformacji, która zaburza strukturę białka i uniemożliwia mu dokończenie katalizy. Zmiana konformacyjna pociąga za sobą przemieszczenie RCL na przeciwległy koniec białka i umieszczenie pomiędzy β-kartką A z wytworzeniem dodatkowej antyrównoległej β-kartki. Przekształca to serpinę ze stanu napiętego do stanu rozluźnionego o mniejszej energii (przejście S w R).
Proteazy serynowe i cysteinowe katalizują rozpad wiązania peptydowego w procesie dwuetapowym. Wpierw reszta katalityczna miejsca aktywnego triady katalitycznej dopuszcza się ataku nukleofilowego na wiązanie peptydowe substratu. Uwalnia się N-koniec z wytworzeniem wiązania estrowego pomiędzy enzymem a substratem. Ten kompleks pomiędzy enzymem a substratem połączonymi kowalencyjnie nazywa się po angielsku acyl-enzyme intermediate. W przypadku zwyczajnych substratów wiązanie estrowe ulega hydrolizie, uwalniając nowy C-koniec i kończąc w ten sposób katalizę. Jednak jeśli proteaza połączy się z serpiną, szybko przechodzi z formy S w formę R, zanim przejściowy kompleks acyl-enzym ulegnie hydrolizie.
Jako że RCL jest kowalencyjnie związane z proteazą wiązaniem estrowym, przejście formy S w R pociąga proteazę ze szczytu w dół serpiny i rozdziela triadę katalityczną. Potraktowana w ten sposób proteaza potrafi jedynie shydrolizować bardzo wolno acyl-enzyme intermediate i dlatego pozostaje związana kowalencyjnie przez dni bądź tygodnie. Serpiny klasyfikuje się jako inhibitory nieodwracalne i samobójcze, gdyż cząsteczka serpiny trwale traci aktywność po związaniu z pojedynczą proteazą, może więc zadziałać tylko raz.


Aktywacja allosteryczna

Zmienność konformacyjna serpin zapewnia im kluczową przewagę nad inhibitorami proteaz działającymi na zasadzie zamka i klucza. W szczególności hamowanie proteaz przez serpiny może podlegać regulacji allosterycznej przez różne kofaktory. Badania krystalograficzne z zastosowaniem promieniowania X antytrombiny, kofaktora heparyny II, MENT i mysiej α1-antychymotrypsyny ujawniły, że serpiny te przyjmują konformację z pierwszymi dwoma resztami aminoacylowymi RCL ułożonymi w szczycie β-kartki A. Częściowo wsunięta konformacja jest ważna, ponieważ kofaktory potrafią ją przekształcić w formę w pełni wyrzuconą. Ta zmienność konformacji czyni serpiny efektywniejszymi inhibitorami.
Przykład stanowi tutaj antytrombina, która krąży w osoczu w postaci częściowo wsuniętej, względnie nieaktywnej. Ułożenie reszty P1 argininy w kierunku serpiny czyni ją nieosiągalną dla proteazy. Po związaniu sekwencji pentasacharydu o dużym powinowactwie znajdujących się w długołańcuchowej heparynie antytrombina przechodzi zmianę konformacyjną. Wyrzuca na zewnątrz RCL i eksponuje arginyl P1. Forma antytrombiny związana z pentasacharydem heparyny stanowi efektywniejszy inhibitor trombiny i aktywnego czynnika X. Co więcej, obie te proteazy związane z koagulacją także zawierają miejsca wiążące heparynę. Heparyna działa tu jako łącznik wiążący obie proteazy i serpinę, w wielkim stopniu przyśpieszając interakcję pomiędzy nimi. Po początkowej interakcji tworzy się ostateczny kompleks serpiny i ugrupowanie heparyny jest usuwane. Jest to interakcja o dużym znaczeniu dla fizjologii. Na przykład po urazie obejmującym ścianę naczynia eksponowana jest heparyna, a antytrombina ulega aktywacji, kontrolując odpowiedź w postaci krzepnięcia. Poznanie podstawy molekularnej tego procesu pozwoliło wprowadzić do lecznictwa nowy lek przeciwkrzepliwy – fondaparynuks, syntetyczną formę pentasacharydu heparyny.
Konformacja latentna

Niektóre serpiny potrafią spontanicznie przechodzić ze stanu S w R bez udziału proteazy, przyjmując konformację zwaną stanem latentnym. Latentne serpiny nie potrafią wchodzić w interakcje z proteazami, nie są już więc ich inhibitorami. Zmiana konformacyjna w kierunku latencji nie jest dokładnie taka sama jak przejście ze stanu S w R związanej serpiny. RCL pozostaje nietknięte, pierwsza nić C-kartki odchyla się, umożliwiając pełne wejście RCL.
Regulacja przejścia latentnego może działać w przypadku niektórych serpin jako mechanizm kontrolny, jest tak np. u PAI-1. Choć białko to tworzone jest w posiadającej zdolność inhibicji konformacji S, ulega autoinaktywacji poprzez zmianę w kierunku stanu latentnego, jeśli nie przywiąże kofaktora – witronektyny. W podobny sposób antytrombina potrafi spontanicznie przejść w stan latentny, co stanowi dodatkowy mechanizm modulacji prócz allosterycznej aktywacji przez heparynę. Dalej N-koniec tengpiny, serpiny z Thermoanaerobacter tengcongensis, jest konieczny dla zatrzymania cząsteczki w jej natywnym stanie o właściwościach inhibicji. Zaburzenie oddziaływań N-końca skutkuje spontaniczną zmianą konformacyjną w kierunku konformacji latentnej.
Zmiana konformacyjna a inne funkcje serpin
Pewne serpiny nie będące infibitorami także wykorzystują zmianę konformacyjną. Na przykład natywna forma S TBG cechuje się wysokim powinowactwem do tyroksyny, natomiast forma R ma małe powinowactwo. Podobnie transkortyna ma wyższe powinowactwo do kortyzolu w swojej natywnej formie S niż w formie R. W tych serpinach odłączenie RCL i przejście S w R powala na przyłączenie ligandu, a nie hamowanie proteaz.
W przypadku niektórych serpin przejście S w R może aktywować procesy sygnalingu. Serpiny takie tworzą kompleks z docelową proteazą, który ulega rozpoznaniu przez receptor. Związanie się z nim rozpoczyna przekaz sygnału. Przejście S w R wobec tego służy do przekazywania komórce informacji o aktywności proteazy. Proces ten różni się od zwyczajnego mechanizmu, w którym serpina wpływa na sygnaling przez hamowanie proteaz zaangażowanych w kaskadę sygnałową.
Degradacja
Hamując docelową proteazę, serpina tworzy z nią trwały kompleks, który wymaga usunięcia. Jeśli chodzi o serpiny zewnątrzkomórkowe, końcowe kompleksy enzym-serpina są szybko usuwane z krwiobiegu. U ssaków w jednym z takich mechanizmów uczestniczy low-density lipoprotein receptor-related protein (LRP1), wiążące kompleksy antytrombiny, PA1-1 i neuroserpiny, powodując endocytozę. Podobnie serpina muszek z rodzaju Drosophila ulega degradacji lizosomalnej po umieszczeniu w komórce przez Lipophorin Receptor-1 (homolog ssaczej rodziny receptorów LDL).
Choroby i serpinopatie
Serpiny biorą udział w znacznym szeregu funkcji fizjologicznych. Mutacje kodujących je genów mogą powodować szereg chorób. Mutacje modyfikujące aktywność, selektywność bądź tendencję do agregacji wpływają na ich działanie. Większość związanych z serpinami chorób stanowi rezultat polimeryzacji serpin i tworzenia agregatów, jednak występuje też kilka innych rodzajów chorób wywołanych ich mutacjami. Niedobór alfa1-antytrypsyny należy do najczęstszych chorób dziedzicznych.
Brak bądź nieaktywność
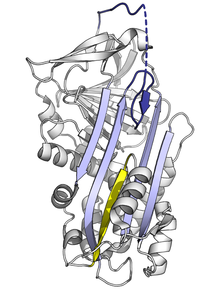
Serpina w stanie naprężonym posiada wysoką energię, a więc mutacje mogą prowadzić do nieprawidłowej zmiany w konformacje o niższej energii, zrelaksowanej bądź latentnej, zanim jeszcze białko zdąży odegrać swą hamującą funkcję.
Mutacje mogą dotykać zasięgu wejścia RCL w kartkę A, co powoduje, że serpina może przejść zmianę konformacyjną S w R przed spotkaniem proteazy. Zmiana taka może zajść w przypadku danej cząsteczki tylko jeden raz. W efekcie zmieniona serpina nie posiada aktywności i zdolności do dokładnej kontroli swego targetu molekularnego. Podobnie mutacje promujące nieprawidłowe przejście w monomeryczny stan latentny powodują chorobę poprzez redukcję ilości aktywnej serpiny. Przykładowo związane z chorobą warianty antytrombiny wibble i wobble sprzyjają przechodzeniu w stan latentny.
Struktura chorobotwórczego mutanta antychymotrypsyny (L55P) wykazuje inną nieaktywną konformację δ. W jej przypadku 4 reszty RCL uległy insercji na szczycie kartki β. Dolna połowa kartki jest wypełniona w efekcie częściowej zmiany jednej z α-helis (hekisy F) do konformacji łańcucha β, z całkowitym wytworzeniem wiązań wodorowych β-kartki. Pozostaje niejasnym, czy inne serpiny mogą przyjmować taką konformację i czy odgrywa ona jakąś rolę w funkcjonowaniu. Spekuluje się, że konformacja δ może być przyjmowana przez globulinę wiążącą tyroksynę podczas uwalniania hormonu. Nieinhibicyjne białka spokrewnione z serpinami także mogą powodować choroby, gdy ulegną mutacji. Na przykład mutacje w SERPINF1 wywołują u człowieka osteogenesis imperfecta typu VI.
Jeśli brakuje potrzebnej serpiny, proteaza normalnie regulowana tym białkiem wykazuje nadmierną aktywność, prowadząc do patologii. W efekcie izolowany niedobór serpiny (np. mutacja typu null) może powodować chorobę. Nokaut genowy wykorzystuje się w badaniach laboratoryjnych szczególnie u myszy w celu ustalenia funkcji serpin i efektów ich braku.
Zmiana specyficzności
W pewnych rzadkich przypadkach zmiana pojedynczej reszty aminokwasowej w RCL zmienia specyficzność serpiny, która reaguje teraz z niewłaściwą proteazą. Na przykład mutacja antytrypsyny Pittsburgh (M358R) powoduje, że α1-antytrypsyna hamuje trombinę, wywołując krwawienia.
Polimeryzacja i agregacja


Większość chorób związanych z serpinami spowodowana jest agregacją białek. Nazywa się je serpinopatiami. Serpiny są podatne na powodujące choroby mutacje promujące tworzenie źle sfałdowanych polimerów z powodu ich inherentnie niestabilnej struktury. Dobrze opisane serpinopatie to niedobór alfa1-antytrypsyny, mogący skutkować rodzinnie występującą rozedmą i czasami marskością wątroby, niektóre rodzinne postacie zakrzepicy związane z niedoborem antytrombiny, dziedziczny obrzęk naczynioruchowy typu 1 bądź 2, związany z niedoborem inhibitora C1, a także FENIB, rodzinna encefalopatia z inkluzjami neuroserpiny, będąca rzadkim rodzajem otępienia, spowodowana polimeryzacją neuroserpiny.
Każdy z monomerów agregatu serpiny przyjmuje nieaktywną, zrelaksowaną konformację z RCL wsunięto w kartkę A. Polimery takie są hiperstabilne w obliczu zmian temperatury, nie hamują proteaz. Nieprawidłowości dotyczące serpin powodują stany patologiczne podobnie jak w przypadku innych proteopatii (jak choćby choroby prionowe). Przebiega to wedle dwóch głównych mechanizmów. Pierwszy polega na braku aktywnej serpiny, co skutkuje niekontrolowaną aktywnością proteaz i niszczeniem tkanek. Drugi obejmuje zaleganie hiperstabilnych polimerów w siateczce śródplazmatycznej, co prowadzi do śmierci komórki i również niszczenia tkanek. W przypadku niedoboru antytrypsyny polimery antytrypsyny powodują śmierć komórek wątroby, niekiedy prowadzi to do uszkodzenia narządu i marskości. W obrębie komórki polimery serpiny są powolnie usuwane poprzez degradację w siateczce śródplazmatycznej. Jednak szczegóły powodowania śmierci komórki przez polimeryzację serpin muszą jeszcze zostać w pełni wyjaśnione.
Uważa się, że fizjologicznie polimery serpina tworzą się poprzez zamianę domen (domain swapping), kiedy to segment jednej serpiny ulega insercji w obrąb innego. Proces taki zachodzi, gdy mutacje bądź czynniki środowiskowe interferują z końcowymi etapami fałdowania się serpin do stanu natywnego, powodując niewłaściwe fałdowanie się wysokoenergetycznych związków pośrednich. Odkryto struktury dimerów i trimerów powstałych w tym procesie. W obrębie dimeru antytrombiny RCL i część kartki A wchodzą w kartkę A innej cząsteczki serpiny. Ttimer antytrypsyny z zamienionymi domenami tworzy się poprzez wymianę całkowicie odmiennych regionów struktury, B-kartki (z każdą pętlą RCL cząsteczki wsuniętą we własną A-kartkę). Zaproponowano również, że serpiny mogą tworzyć struktury o zamienionych domenach przez wsunięcie RCL jednego białka do kartki A innego (polimeryzacja kartki A). Struktury dimeru i trimeru uważane są za bloki budulcowe chorobotwórczych agregatów, ale dokładny mechanizm tego zjawiska pozostaje niejasny.
Strategie terapeutyczne
Kilka podejść terapeutycznych wykorzystano bądź poddano badaniu w celu leczenia najczęstszej z serpinopatii: niedoboru antytrypsyny. Augmentacja antytrypsyny zaakceptowana została w leczeniu rozedmy płuc związanej z ciężkim niedoborem antytrypsyny. W tej terapii antytrypsynę uzyskuje się z osocza dawców krwi, po czym podaje dożylnie. Preparat sprzedawany jest jako Prolastin. W leczeniu stanów związanych z ciężkim niedoborem antytrypsyny efektywne okazały się przeszczepy płuc i wątroby. W modelach zwierzęcych wykorzystywano z powodzeniem technikę gene targeting w indukowanych pluripotencjalnych komórkach macierzystych, korygując defekt polimeryzacji antytrypsyny i przywracając zdolność wątroby ssaka do wydzielania aktywnej antytrypsyny. Wynaleziono też małe cząsteczki blokujące polimeryzację antytrypsyny in vitro.
Ewolucja
Serpiny stanowią najszerzej rozpowszechnioną i największą nadrodziną inhibitorów białkowych. Pierwotnie sądzone, że ograniczają się tylko do organizmów eukariotycznych. Później jednak znaleziono je u bakterii, archeonów, a nawet niektórych wirusów. Pozostaje niejasne, czy geny prokariotyczne są potomkami genów pierwotnych serpin prokariotycznych, czy też efektem horyzontalnego transferu genów z eukariotów. Większość serpin wewnątrzkomórkowych należy do jednego kladu bez względu na pochodzenie roślinne bądź zwierzęce. Wskazuje to na podział serpin na wewnątrzkomórkowe i zewnątrzkomórkowe jeszcze przed podziałem na rośliny i zwierzęta. Wyjątki obejmują wewnątrzkomórkową serpinę szoku cieplnego HSP47, będącą szaperonem kluczowym dla właściwego fałdowania się kolagenu oraz cyklu zachodzącego pomiędzy częścią cis aparatu Golgiego i siateczką śródplazmatyczną.
Hamowanie proteaz uznaje się za pierwotną funkcję. Niebędące inhibitorami serpiny są efektem neofunkcjonalizacji. Zmiana konformacyjna S w R została także wykorzystana przez pewne serpiny do regulacji powinowactwa do ich targetów molekularnych.
Rozpowszechnienie
Zwierzęta
Człowiek
Genom ludzki zawiera 16 kladów serpin, oznaczanych literami od A do P, w tym 29 inhibitorów i 7 serpin niebędących inhibitorami białek. System nazewnictwa serpin ludzkich bazuje na analizie filogenetycznej około 500 serpin z 2001, stosującej nazwy typu serpinXY, gdzie X oznacza klad białek, a Y jest numerem białka w obrębie danego kladu. Funkcję serpin człowieka ustalono dzięki połączeniu badań biochemii, ludzkich chorób genetycznych i mysich modeli stosujących knockout.
| Nazwa genu | Nazwa popularna | Lokalizacja | Function / Activity | Efekt niedoboru | Choroba człowieka | Miejsce na chromosomie | |
|---|---|---|---|---|---|---|---|
| SERPINA1 | α1-antytrypsyna | Zewnątrzkomórkowa | Inhibitor elastazy neutrofili. C-koniec może hamować infekcję HIV-1. | niedobór alfa1-antytrypsyny objawia się rozedmą, polimeryzacja w wątrobie – marskością. | 14q32.1 | ||
| SERPINA2 | Antitrypsin-related protein | Zewnątrzkomórkowa | Prawdopodobnie pseudogen. | 14q32.1 | |||
| SERPINA3 | α1-antychymotrypsyna | Zewnątrzkomórkowa | Inhibitor katepsyny G, pełni dodatkową rolę w kondensacji chromatyny w komórkach wątroby. | Dysregulacja w chorobie Alzheimera. | 14q32.1 | ||
| SERPINA4 | Kalistatyna | Zewnątrzkomórkowa | Inhibitor kalikreiny, regulatora funkcji naczyniowych. | Niedobór u szczurów z nadciśnieniem zaostrza uszkodzenie nerek i układu sercowo-naczyniowego. | 14q32.1 | ||
| SERPINA5 | inhibitor białka C | Zewnątrzkomórkowa | Inhibitor aktywnego białka C. Wewnątrzkomórkowo ochrona przed fagocytozą bakterii. | Knockout u samców myszy powoduje bezpłodność. Akumulacja w altywnych plakach w SM. | 14q32.1 | ||
| SERPINA6 | Transkortyna | Zewnątrzkomórkowa | Wiązanie kortyzolu. | Niedobór związany z przelekłym zmęczeniem. | 14q32.1 | ||
| SERPINA7 | Globulina wiążąca tyroksynę | Zewnątrzkomórkowa | Wiązanie tyroksyny. | Niedobór powoduje objawy niedoczynności tarczycy. | Xq22.2 | ||
| SERPINA8 | Angiotensynogen | Zewnątrzkomórkowa | Rozkładany przez reninę tworzy angiotensynę I | Knockout u myszy powoduje nadciśnienie. | Istnieją warianty związane z nadciśnieniem. | 1q42-q43 | |
| SERPINA9 | Centeryna / GCET1 | Zewnątrzkomórkowa | Inhibitor utrzymujące dziewicze limfocyty B. | Silna ekspresja w większości chłoniaków B-komórkowych. | 14q32.1 | ||
| SERPINA10 | Inhibitor proteaz związany z białkiem Z | Zewnątrzkomórkowa | Wiąże białko Z, inaktywuje czynnik Xa i czynnik XIa | 14q32.1 | |||
| SERPINA11 | – | Prawdopodobnie zewnątrzkomórkowa | Nieznana | 14q32.13 | |||
| SERPINA12 | waspina | Zewnątrzkomórkowa | Inhibitor kallikreiny 7, adipokina uczulająca na insulinę. | Wysokie stężenie w osoczu wiąże się z cukrzycą typu 2. | 14q32.1 | ||
| SERPINA13 | – | Prawdopodobnie zewnątrzkomórkowa | Nieznana | 14q32 | |||
| SERPINB1 | Inhibitor elastazy neutrofilowej | Wewnątrzkomórkowa | Inhibitor elastazy neutrofilowej. | Knockout u myszy powoduje defekt przeżywania neutrofili i niedobór immunologiczny. | 6p25 | ||
| SERPINB2 | inhibitor 2 aktywatora plazminogenu | Wewnątrz- i zewnątrzkomórkowa | Inhibitor zewnątrzkomórkowego uPA. Funkcja wewnątrzkomórkowa niejasna, może chronić przed infekcją wirusową. | Niedobór u myszy redukuje odpowiedź na infestację niecieniami. Knockout u myszy nie przejawia się widocznym fenotypem. | 18q21.3 | ||
| SERPINB3 | Squamous cell carcinoma antigen-1 (SCCA-1) | Wewnątrzkomórkowa | Inhibitor proteaz cysteinowych typu papainy oraz kaepsyn K, L and S | Knockout mysiej Serpinb3a (homologu ludzkich SERPINB3 i SERPINB4) zmniejsza produkcję śluzu w mysim modelu astmy. | 18q21.3 | ||
| SERPINB4 | Squamous cell carcinoma antigen-2 (SCCA-2) | Wewnątrzkomórkowa | Inhibitor proteaz serynowych typu chymotrypsyny, katepsyny G i chymazy. | Knockout mysiej Serpinb3a (homologu ludzkich SERPINB3 i SERPINB4) zmniejsza produkcję śluzu w mysim modelu astmy. | 18q21.3 | ||
| SERPINB5 | maspina | Wewnątrzkomórkowa | Nieznana, nie jest inhibitorem | Knockout u myszy pierwotnie uznano za letalny, ale okazał nie przejawiać się ewidentnym fenotypem. Ekspresja może bbyć wskaźnikiem prognostycznym ekspresji sąsiedniego genu supresorowego (fosfatazy PHLPP1). | 18q21.3 | ||
| SERPINB6 | PI-6 | Wewnątrzkomórkowa | Inhibitor katepsyny G | Knockout u myszy powoduje utratę słuchu i łagodną neutropenię. | Niedobór związany z utratą słuchu. | 6p25 | |
| SERPINB7 | Megsin | Wewnątrzkomórkowa | Bierze udział w dojrzewaniu megakariocytów. | Nadmierna ekspresja u myszy powoduje chorobę nerek. Knockout u myszy nie powoduje nieprawidłowości histologicznych. | Mutacje związane z keratozą dłoni i stóp typu Nagashima. | 18q21.3 | |
| SERPINB8 | PI-8 | Wewnątrzkomórkowa | Możliwy inhibitor furyny. | 18q21.3 | |||
| SERPINB9 | PI-9 | Wewnątrzkomórkowa | Inhibitor cytotoksycznej proteazy granzymu B | Knockout u myszy wywołuje dysfunkcję immunologiczną. | 6p25 | ||
| SERPINB10 | bomapina | Wewnątrzkomórkowa | Nieznana | Knockout u myszy nie wywołuje oczywistego fenotypu (C57/BL6; szczep laboratoryjny BC069938). | 18q21.3 | ||
| SERPINB11 | Wewnątrzkomórkowa | Nieznana | Mysia Serpinb11 jest aktywnym inhibitorem, podczas gdy ludzki ortolog jest nieaktywny. Niedobór u kuców wiąże się z chorobą kopyt. | 18q21.3 | |||
| SERPINB12 | Yukopin | Wewnątrzkomórkowa | Nieznana | 18q21.3 | |||
| SERPINB13 | Hurpin/Headpin | Wewnątrzkomórkowa | Inhibitor proteaz cysteinowych przypominających papainę. | 18q21.3 | |||
| SERPINC1 | Antytrombina | Zewnątrzkomórkowa | Inhibitor krzepnięcia, zwłaszcza czynników X, IX i trombiny. | Knockout u myszy jest letalny. | Niedobór powoduje zakrzepicą i innymi zaburzeniami krzepnięcia (serpinopatiami). | 1q23-q21 | |
| SERPIND1 | kofaktor II heparyny | Zewnątrzkomórkowa | Inhibitor trombiny. | Knockout u myszy jest letalny. | 22q11 | ||
| SERPINE1 | inhibitor aktywatora plazminogenu | Zewnątrzkomórkowa | Inhibitor trombiny, uPA i TPa. | 7q21.3-q22 | |||
| SERPINE2 | Glia derived nexin / Protease nexin I | Zewnątrzkomórkowa | Inhibitor uPA i tPA. | Nieprawidłowa ekspresja prowadzi do niepłodności męskiej. Knockout u myszy wywołuje padaczkę. | 2q33-q35 | ||
| SERPINF1 | Pigment epithelium derived factor (PEDF) | Zewnątrzkomórkowa | Nieinhibicyjna, możliwe że chodzi o cząsteczkę antyangiogenną. PEDF wiąże haluronian. | Knockout u myszy wpływa na unaczynienie i masę trzustki i stercza. Promuje zależną od Notch odnowę dojrzałych przykomorowych komórek macierzystych. Mutacje u ludzi powodują wrodzoną łamliwość kości typu VI. | 17p13.3 | ||
| SERPINF2 | α2-antiplasmin | Zewnątrzkomórkowa | Inhibitor plazminy i fibrynolizy. | Knockouts u myszy powoduje wzrost fibrynolizy, jednak bez zaburzeń krwawienia. | Niedobór wywołuje rzadkie zaburzenie krwawienia. | 17pter-p12 | |
| SERPING1 | Inhibitor C1 | Zewnątrzkomórkowa | Inhibitor esterazy C1. | Kilka polimorfizmów związanych ze zwyrodnieniem plamki żółtej i wrodzonym obrzękiem naczyniowym. | 11q11-q13.1 | ||
| SERPINH1 | HSP47 | Wewnątrzkomórkowa | Nieinhibicyjna. Chaperon ułatwiający fałdowanie się kolagenu. | Knockouts u myszy powoduje zgon. | Mutacje u ludzi powodują ciężką wrodzoną łamliwość kości. | 11p15 | |
| SERPINI1 | Neuroserpina | Zewnątrzkomórkowa | Inhibitor tPA, uPA i plazminy. | Mutacja powoduje rodzinną encefalopatię z inkluzjami ciałek neuroserpiny, przebiegającą z otępieniem. | 3q26 | ||
| SERPINI2 | Pancpin | Zewnątrzkomórkowa | Nieznana | Niedobór u myszy powoduje niedoczynność zewnątrzwydzielniczą trzustki przez utratę komórek gronek | 3q26 |
Wyspecjalizowane serpiny ssaków
W przypadków wielu serpin ssaków nie znaleziono oczywistych ortologów u człowieka. Na przykład liczne serpiny gryzoni (szczególnie niektóre mysie serpiny wewnątrzkomórkowe) jak też serpiny maciczne – są to serpiny należące do kladu serpin A, kodowane przez gen SERPINA14, produkowane przez endometrium ograniczonej grupy ssaków z kladu Laurasiatheria pod wpływem progesteronu lub estrogenów. Nie są to prawdopodobnie funkcjonalne inhibitory białkowe, mogą funkcjonować w ciąży, hamując odpowiedź odpornościową matki przeciw zarodkowi lub uczestniczyć w transporcie przezłożyskowym.
Owady
Genom Drosophila melanogaster zawiera 29 genów kodujących serpiny. Analiza sekwencji aminokwasowej umieściła 14 z tych serpin w kladzie Q, a 3 w kladzie K. Pozostałych 12 sierocych serpin nie należy do żadnego kladu serpin. Kladystyczny system klasyfikacji serpin sprawia trudności przy zastosowaniu do serpin Drosophila i zamiast niego zaadaptowano system nomenklaturowy bazujący na ulokowaniu ich genów na chromosomach wywilżny. 13 z genów serpin Drosophila występuje jako izolowane geny w genomie (w tym Serpin-27A), pozostałych 16 zorganizowanych jest w 3 klastery leżące na chromosomach w miejscach 28D (2 serpiny), 42D (5), 43A (4), 77B (3) i 88E (2).
Badania serpin Drosophila ujawniła, że serpina 27A hamuje proteazę Easter (końcową proteazę kaskad proteolitycznych Nudel, Gastrulation Defective, Snake i Easter) i w ten sposób kontroluje wzorzec osi grzbietowo-brzusznej. Easter działa poprzez rozkład wiążącego chemokiny ligandu Spätzle, co skutkuje sygnalizacją mediowaną przez receptory toll-podobne. Odgrywając ważną rolę w rozwoju embrionalnym, sygnaling toll jest także ważny we wrodzonej odpowiedzi immunologicznej owadów. W związku z tym serpina 27A kontroluje również owadzią odpowiedź immunologiczną. U chrząszcza mącznika młynarka białko SPN93 zawiera 2 odrębne domeny serpinowe regulujące kaskadę proteolityczną toll.
Nicienie
Genom nicienia Caenorhabditis elegans koduje 9 serpin, z których żadna nie ma sekwencji sygnałowej, w związku z czym są prawdopodobnie wewnątrzkomórkowe. Jednak tylko 5 z nich wydaje się funkcjonować jako inhibitory proteaz. Wśród nich SRP-6 pełni funkcję ochronną i przeciwdziała indukowanemu stresem związanemu z kalpainą rozpadowi lizosomów. Co więcej, SRP-6 hamuje lizosomalną proteazę cysteinową wydzielaną po przerwaniu ciągłości lizosomów. W związku z tym nicienie pozbawione SRP-6 są wrażliwe na stres. W szczególności nicienie z knockoutem SRP-6 umierają umieszczone w wodzie (letalny fenotyp stresu hipoosmotycznego, Osl). Zasugerowano, że izosomy odgrywają główną rolę kontrolującą w determinacji przeznaczenia komórek.
Rośliny
Serpiny roślin należały do pierwszych zidentyfikowanych członków swej nadrodziny. Białko Z jęczmienia występuje bardzo obficie w jęczmiennym ziarnie i jest jednym z głównych białek piwa. Genom rośliny modelowej rzodkiewnika pospolitego zawiera 18 genów kojarzących się z serpinami, jednak tylko 8 z nich obejmuje pełnej długości sekwencje kodujące serpiny.
Serpiny roślin są potężnymi inhibitora ssaczych proteaz podobnych do chmotrypsyny in vitro, najlepiej przebadano jęczmienną serpinę Zx (BSZx), zdolną do hamowania trypsyny i chymotrypsyny, jak też kilku czynników krzepnięcia. Jednak bliscy krewni chymotrypsynopodobnych proteaz serynowych nie występują u roślin. RCL kilku serpin z ziaren pszenicy i żyta zawiera powtarzalne sekwencje poli-Q podobne do obecnych w prolaminach bielma. Zasugerowano, że serpiny roślinne mogą działać poprzez hamowanie proteaz owadów bądź mikroorganizmów, które inaczej trawiłyby białka zapasowe ziarna. Hipotezę tą wspiera identyfikacja specyficznych serpin roślinnych w sokach łyka dyni (CmPS-1) i ogórka. Choć dostrzeżono odwrotną korelację pomiędzy regulacją w górę ekspresji CmPS-1 i przeżyciem mszyc, badania in vitro ujawniły, że rekombinant CmPS-1 nie wydaje się wpływać na przeżycie owadów.
Proponowano alternatywne funkcje i proteazy docelowe serpin roślinnych. Serpina rzodkiewnika AtSerpin1 (At1g47710; 3LE2) mediuje punkt kontrolny programowanej śmierci komórki, wpływając na proteazę cysteinową papainopodobną 'Responsive to Desiccation-21' (RD21). AtSerpin1 hamuje także przypominające metakaspazę białka in vitro. Dwie inne serpiny Arabidopsis, AtSRP2 (At2g14540) i AtSRP3 (At1g64030), wydają się uczestniczyć w odpowiedzi na uszkodzenie DNA.
Grzyby
Dotychczas określono pojedynczą serpinę grzybów: celpinę z Piromyces, ze szczepu E2. Piromyces to rodzaj beztlenowych grzybów spotykanych w jelitach przeżuwaczy, ważny w trawieniu pokarmu roślinnego. Przewiduje się, że celpina będzie inhibitorem, zawierając dwie N-końcowe domeny dokerynowe prócz domen serpinowych. Dokeryny spotyka się często wśród białek lokalizujących się w cellulosomie grzybowym, dużym wewnątrzkomórkowym kompleksie złożonym z wielu białek, prowadzącym rozkład celulozy. Zasugerowano więc, że celpina może chronić celulosom przed proteazami roślinnymi. Pewne serpiny bakteryjne lokalizują się podobnie jak cellulosom.
Prokarioty
Przewidywane geny serpin są sporadycznie rozmieszczone u prokariotów. Badania in vitro niektórych z tych cząsteczek ujawniły, że mają one zdolność hamowania proteaz, sugeruje się, że działają jako ich inhibitory in vivo. Pewne serpiny prokariotyczne spotyka się u ekstremofili. W związku z tym i w kontraście w stosunku do serpin ssaczych cząsteczki te posiadają podwyższoną oporność na denaturację spowodowaną ciepłem. Dokładna rola większości serpin bakteryjnych pozostaje nieznana, choć serpiny Clostridium thermocellum lokalizują się w cellulosomie. Zasugerowano, że rola serpin związanych z cellulosomem może polegać na ochronie przed niepożądaną aktywności proteaz przeciwko cellusomowi.
Wirusy
Serpiny podlegają ekspresji także u wirusów, jako środek uniknięciu odpowiedzi immunologicznej gospodarza. W szczególności serpiny wytwarzają pokswirusy, w tym wirus krowianki i Orthopoxvirus. Stały się one obiektami zainteresowania z powodu możliwości wykorzystania w terapii zaburzeń immunologicznych i chorób zakaźnych, jak też w transplantologii. Serp1 hamuje toll-zależną wrodzoną odpowiedź immunologiczną i umożliwia nieograniczone przeżycie alloprzeszczepu serca u szczurów. Crma i Serp2 są międzyklasowymi inhibitorami i atakują zarówno proteazy serynowe (granzym B, choć słabo), jak i cysteinowe (kaspazy 1 i 8). W porównaniu do ich ssaczych odpowiedników serpiny wirusowe zawierają znaczące delecje elementów struktury drugorzędowej. Konkretnie crmA brakuje D-helisy, jak też znacznych części helis A i E.