
| morbus Behçeti | |
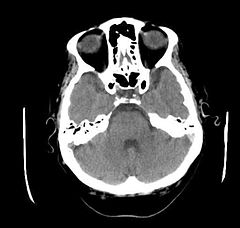 CT głowy pacjenta z objawami neurologicznymi w przebiegu choroby Behçeta, hiperdensyjna lewa zatoka esowata | |
| Klasyfikacje | |
| ICD-10 |
M35.2 |
|---|---|


Choroba Behçeta (choroba Adamantiadesa-Behçeta, łac. morbus Behçeti, ang. Behçet's disease) – układowe zapalenie naczyń małych, średnich i dużych, które współistnieje z bolesnymi owrzodzeniami skóry oraz błon śluzowych narządów płciowych i jamy ustnej, a także z zajęciem procesem chorobowym różnych narządów (stawy, oczy, układ nerwowy, przewód pokarmowy).
Historia
Choroba po raz pierwszy została prawdopodobnie opisana już w V wieku p.n.e. przez Hipokratesa. Autorem pierwszego opisu schorzenia w czasach nowożytnych był w roku 1931 grecki okulista Benediktos Adamantiades. Choroba przyjęła natomiast nazwę od nazwiska Hulusi Behçeta (czyt. /behˈtʃeta/; 1889–1948), tureckiego dermatologa, który w roku 1924 stwierdził objawy tego schorzenia u jednego ze swoich pacjentów i opisał je jako symptomy nowej jednostki chorobowej w 1936 roku w Archives of Skin and Veneral Diseases. Nazwa choroby została oficjalnie przyjęta na Międzynarodowym Kongresie Dermatologicznym w Genewie w roku 1947.
Epidemiologia
Choroba występuje najczęściej w Turcji (80–370 przypadków/100 tys. mieszkańców/rok) i krajach Bliskiego Wschodu. W Europie i USA jest to schorzenie zdecydowanie rzadsze, stwierdza się 0,1-7,5 nowych przypadków na 100 tys. obywateli w ciągu roku. Chorują najczęściej ludzie w 3. dekadzie życia a zapadalność jest podobna u obu płci, poza krajami Bliskiego Wschodu, gdzie choroba dotyczy głównie mężczyzn.
Etiologia
Przyczyna choroby nie jest znana, choć bierze się pod uwagę zaburzenia układu immunologicznego, które prowadzą do zapalenia naczyń.
Kryteria rozpoznania
- bolesne afty w jamie ustnej (92-100%), pojawiające się co najmniej 3 razy w ciągu roku
- obecność co najmniej 2 z poniższych cech:
- owrzodzenia narządów płciowych (57-93%)
- zmiany dotyczące oczu (29-100%): zapalenie błony naczyniowej oka lub zapalenie naczyń siatkówki oka
- zmiany skórne (38-99%) takie jak: rumień guzowaty, zmiany pseudo-trądzikowe, wędrujące zapalenie żył powierzchownych skóry
- patergia, czyli powstanie zmian zapalnych dotyczących skóry po jej niewielkim urazie
- zapalenie stawów bez obecności nadżerek (16-84%)
- zaburzenia ośrodkowego układu nerwowego – bóle głowy, zaburzony nastrój, uszkodzenie różnych struktur mózgu (tzw. postać „neuro-Behçet”)
- zakrzepowe zapalenie żył głębokich
- zaburzenia układu pokarmowego – biegunka
Nowe kryteria rozpoznania choroby Behceta - ChB (ICBD - International Criteria for Behcet's Disease) wprowadzono w roku 2013. Są one zarekomendowane jako obowiązujące w chwili obecnej (2018). W odróżnieniu od kryteriów z roku 1990 (ISG - International Study Group) opisanych powyżej, występowanie aft w jamie ustnej nie jest dłużej kryterium obowiązkowym. Kryterium patergii jest kryterium dodatkowym, opcjonalnym. Oprócz pięciu „starych” kryteriów (jama ustna, genitalia, skóra, oczy, patergia), wprowadzono dwa nowe: kryterium układu nerwowego i kryterium układu krwionośnego. Diagnozowanie choroby odbywa się metodą systemu punktowego. Za chorego na ChB uznaje się osobę uzyskującą 4 punkty lub więcej. 2 punkty przypisane są trzem kryteriom - aftozie jamy ustnej, aftozie genitaliów, oraz problemom związanym z oczami. Jeden punkt przypisany jest zmianom skórnym, zmianom w układzie nerwowym oraz zmianom w układzie krwionośnym. Dodatkowo można otrzymać 1 punkt za pozytywny wynik testu patergii. Uzyskanie sześciu lub więcej punktów klasyfikuje pacjenta jako chorego z prawdopodobieństwem w granicach 99 procent (prawdopodobieństwo że osoba zdrowa zostanie zakwalifikowana jako chora jest mniejsze niż 1%) Jakakolwiek kombinacja dwóch z trzech kryteriów za 2 punkty klasyfikuje pacjenta jako osobę chorą, jest to znaczne obniżenie poziomu rozpoznawalności ChB względem ISG, gdzie pacjent musiał spełniać 3 kryteria (główne - jama ustna, plus dwa z czterech dodatkowych).
ICBD różni od ISG znaczny wzrost czułości testu - z 81% do 94%, przy jednoczesnym niewielkim spadku swoistości z 96% do 92%, co jest nadal wynikiem zadowalającym. Również próba użyta do wnioskowania jest bardziej reprezentatywna - przeprowadzono badania na 2556 pacjentach z ChB oraz 1163 pacjentach kontrolnych z 27 krajów, natomiast ISG obejmowało dane od 914 pacjentów z 7 krajów (jedynie Iran, Turcja, Japonia, Tunezja, Wielka Brytania, USA i Francja). Test patergii uznano za opcjonalny, gdyż jego czułość dla pacjentów z basenu Morza Śródziemnego była bardzo wysoka - w granicach 95%, natomiast u pacjentów z Europy i USA była znacznie niższa. Badania były prowadzone w latach 2005-2006.
| Objaw | Liczba pacjentów Z ChB | Czułość % | Liczba pacjentów naśladowczych | Swoistość % |
|---|---|---|---|---|
| Aftoza jamy ustnej | 1248 | 98 | 378 | 35 |
| Aftoza genitaliów | 950 | 74 | 35 | 94 |
| Manifestacje skórne | 899 | 70 | 76 | 87 |
| Psedofolliculitis | 674 | 53 | 48 | 92 |
| Rumień guzowaty | 406 | 32 | 27 | 95 |
| Aftoza skóry | 56 | 4 | 10 | 98 |
| Zmiany w oku | 703 | 55 | 139 | 76 |
| Zapalenie błony naczyniowej przednie (anterior uveitis) | 508 | 40 | 120 | 79 |
| Zapalenie błony naczyniowej tylne (posterior uveitis) | 482 | 38 | 76 | 87 |
| Zapalenie ukrwienia siatkówki (retinal vasculitis) | 288 | 23 | 21 | 96 |
| Manifestacje stawowe | 652 | 51 | 213 | 63 |
| Arthralgia | 443 | 35 | 161 | 72 |
| Arthritis | 289 | 23 | 76 | 87 |
| Manifestacje neurologiczne | 212 | 17 | 16 | 97 |
| OUN | 155 | 12 | 9 | 98 |
| Obwodowy układ nerwowy | 63 | 5 | 8 | 99 |
| Objawy układu krwionośnego | 242 | 19 | 36 | 94 |
| Objawy układu pokarmowego | 80 | 6 | 17 | 97 |
| Zapalenie najądrza (epididymitis) | 91 | 7 | 9 | 98 |
| Pozytywny test patergii | 419 | 47 | 33 | 92 |
Leczenie
Obecna terapia polega na łagodzeniu objawów chorobowych, zmniejszeniu stanu zapalnego oraz kontroli układu odpornościowego pacjenta. W leczeniu zmian skórnych wykorzystuje się glikokortykosteroidy, takrolimus, kolchicynę, dapson, antybiotyki, talidomid, interferon-α, metotreksat. W terapii zmian narządowych stosuje się glikokortykosteroidy, intereferon-α, cyklosporynę, azatioprynę, cyklofosfamid. W ostatnich latach wprowadza się do leczenia z dobrym skutkiem nowoczesne leki biologiczne skierowane przeciwko TNF-α: infliksimab zatwierdzony do terapii zapalenia błony naczyniowej oka czy etanercept w leczeniu zmian skórnych i śluzówkowych.
Rokowanie
Na ogół dobre, o ile nie dojdzie do zajęcia ośrodkowego układu nerwowego lub płuc.
Bibliografia
- P.B. Dimakakos, B. Tsiligiris, T. Kotsis, The physician B. Adamantiades and his contribution to the disease Adamantiades-Behçet, „Int Angiol”, 18 (2), International Angiology, 1999, s. 176-81, PMID: 10424377 (ang.).
- A. Eguia, M. Villarroel, R. Martínez-Conde, M.A. Echebarría i inni. Adamantiades-Behçet disease: an enigmatic process with oral manifestations. „Medicina Oral, Patología Oral y Cirugía Bucal”. 11(1), s. E6-11, 1 2006. PMID: 16388284. (ang.).
- Behcets Disease. Johns Hopkins Vasculitis Center. [dostęp 2014-05-10]. (ang.).
- V. Kontogiannis, R.J. Powell. Behçet's disease. „Postgraduate Medical Journal”. 76(900), s. 629-37, 10 2000. PMID: 11009577. (ang.).
- M. Melikoglu i inni, Short-term trial of etanercept in Behcet's disease: a double blind, placebo controlled study, „Journal of Rheumatology”, 32 (1), 2005, s. 98-105, PMID: 15630733 (ang.).
- T. Saylan. Life Story of Dr. Hulusi Behçet. „Yonsei Medical Journal”. 38(6), s. 327-332, 12 1997. PMID: 9509901. (ang.).
- P.P. Sfikakis, P.G. Theodossiadis, C.G. Katsiari, P. Kaklamanis i inni. Effect of infliximab on sight-threatening panuveitis in Behcet's disease. „Lancet”. 358 (9278), s. 295-6, 2001. PMID: 11498218. (ang.).
- P.P. Sfikakis. Behcet's disease: a new target for anti-tumour necrosis factor treatment. „Annals of the Rheumatic Diseases”. 61 supl.2, s. ii51-3, 2002. PMID: 12379622. (ang.).
- Andrzej Szczeklik: Choroby wewnętrzne. T. 2.. Kraków: Medycyna Praktyczna, 2006, s. 1700. ISBN 83-7430-031-0.
- C.C. Zouboulis. Epidemiology of Adamantiades-Behçet's disease. „Annales de Médecine Interne”. 150(6), s. 488-98, 10 1999. PMID: 10615535. (fr.).
Linki zewnętrzne
- Polskie stowarzyszenie Chorych na Układowe Zapalenia Naczyń "Vasculitis" : Stowarzyszenie Chorych na Układowe Zapalenia Naczyń "Vasculitis" (pol.)
- Zapalenie naczyń w chorobie Behceta. w bazie stowarzyszenia Vasculitis (pol.)