
| morbus von Recklinghausen | |
| |
| Klasyfikacje | |
| ICD-10 | |
|---|---|

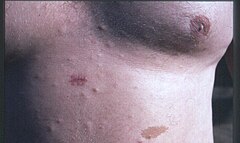
Nerwiakowłókniakowatość typu 1 (neurofibromatoza typu 1, choroba von Recklinghausena, zespół Recklinghausena, ang. neurofibromatosis type I, NF1) – choroba genetyczna o dziedziczeniu autosomalnym dominującym, należąca do grupy fakomatoz. W obrazie klinicznym choroby występują zmiany skórne, oczne, guzy wewnątrzczaszkowe i inne nowotwory o lokalizacji pozaczaszkowej, a także zmiany kostne. Ze względu na dużą zmienność objawów klinicznych nierzadko rozpoznanie choroby jest opóźnione w przypadkach o łagodnej ekspresji. Nerwiakowłókniakowatość typu 1 spowodowana jest mutacją w genie NF1 kodującym neurofibrominę 1. Choroba ta jest nieuleczalna.
Mimo iż synonimem innego schorzenia – dystrofii kości przytarczyczkowej, także jest określenie choroba Recklinghausena nie należy mylić go z nerwiakowłokniakowatością typu 1.
Historia
Pierwszy opis choroby przedstawił irlandzki chirurg Robert William Smith w 1849. Klasyczna praca Friedricha Daniela von Recklinghausena z 1882 roku zawierała opis symptomatologii choroby i wprowadziła do medycyny termin neurofibromatosis; do dziś nerwiakowłókniakowatość typu 1 określa się niekiedy jako chorobę von Recklinghausena.
Etiologia
Choroba spowodowana jest odziedziczoną mutacją genu supresorowego NF1 w locus 17q11.2 kodującego białko neurofibrominę 1. Ponieważ gen NF1 ma duże rozmiary (ponad 350 tys. par zasad), to w ok. połowie przypadków schorzenie jest wynikiem nowej mutacji i nie ma charakteru rodzinnego. Wskutek spontanicznej mutacji drugiego allela genu NF1 i tzw. utraty heterozygotyczności rozwijają się charakterystyczne dla choroby guzy nowotworowe i hamartomatyczne.
Stwierdzono, że homozygotyczność mutacji konstytucyjnych w jednym z genów odpowiedzialnych za mechanizm naprawy DNA przez wycinanie niesparowanych nukleotydów (mismatch repair, MMR), MLH1, MSH2, MSH6 i PMS2 predysponuje do łagodnej postaci neurofibromatozy typu 1, schorzeń hematologicznych i guzów mózgu. U heterozygot mających konstytucyjne mutacje w tych genach rozwija się HNPCC. Zasugerowano, że zespół spowodowany bialleliczną mutacją genów MMR stanowi odrębną jednostkę chorobową, na której określenie zaproponowano nazwy zespołu nowotworów wieku dziecięcego (childhood cancer syndrome, CCR), zespołu niedostatecznej naprawy niesparowanych nukleotydów (mismatch repair deficiency syndrome, MMR-D) albo zespołu CoLoN (Colon tumours and/or leukaemia/lymphoma and/or neurofibromatosis features).
Epidemiologia
NF1 jest stosunkowo częstą chorobą genetyczną i najczęstszą z fakomatoz; szacuje się, że częstość choroby wynosi około 1:3500 żywych urodzeń i przynajmniej 1:4000-1:5000 w populacji.
Objawy i przebieg




NF1 charakteryzuje się różnorodnymi zmianami w tkankach pochodzenia ektodermalnego. Objawy w NF1 można podzielić na duże i małe.
Niekiedy choroba pozostaje nierozpoznana, co jest spowodowane z jednej strony dyskretnym przebiegiem tego schorzenia, a także tym że, nasilenie objawów wykazuje bardzo duże spektrum, zarówno wśród członków rodziny jak i także w ciągu życia tego samego pacjenta.
Objawy duże:
- plamy café au lait (>99%), czyli barwy kawy z mlekiem, rozmieszczone są na całym ciele, niekiedy już od urodzenia, zazwyczaj pojawiają się w okresie niemowlęcym;
- piegowate nakrapiania średnicy 2–3 mm i przebarwienia skórne (70%) w okolicach pachowych i pachwinowych (objaw Crowe'a), pojawiają się najczęściej w okresie dojrzewania;
- guzki podskórne będące histologicznie nerwiakowłókniakami (>99%)
- guzki Lischa (90-95%), ciemnożółte lub brązowawe hamartomatyczne guzki tęczówki, dobrze widoczne w lampie szczelinowej.
Objawy małe:
- makrocefalia (40-50%)
- niedobór wzrostu (30%) wynikający z podwzgórzowej lokalizacji guzów.
Objawy wtórne i powikłania:
- nerwiakowłókniaki splotowate (35%), o różnych lokalizacjach (tkanka podskórna, narządy wewnętrzne)
- niepełnosprawność intelektualna, deficyty psychospołeczne, nadpobudliwość, drobne nieprawidłowości orientacji wzrokowo-przestrzennej (30%)
- padaczka (5%), zwykle pod postacią napadów częściowych złożonych lub uogólnionych napadów toniczno-klonicznych;
- guzy ośrodkowego układu nerwowego:
- glejak nerwu wzrokowego (1,5%)
- nerwiakowłókniaki rdzenia kręgowego
- glejak ze stenozą wodociągu mózgu (1,5%)
- osłoniak nerwu przedsionkowego - Schwannoma nerwu przedsionkowo-ślimakowego
- nowotwory złośliwe:
- złośliwe guzy otoczki nerwów obwodowych, MPNST (1,5%; ryzyko że u pacjenta w ciągu całego życia rozwiną się te guzy, wynosi 7-12%)
- mięsaki prążkowanokomórkowe (1,5%)
- guz chromochłonny (0,7%)
- białaczki, zwłaszcza wczesnodziecięca białaczka nielimfocytowa (<1,0%)
- rakowiak dwunastnicy (1,5%)
- powikłania ortopedyczne: dysplazje i deformacje kostne, zwłaszcza skolioza odcinka piersiowego kręgosłupa, dysplazja skrzydeł większych kości klinowej, deformacje kości strzałkowej i piszczelowej, złamania patologiczne z tendencją do tworzenia stawów rzekomych (25%)
- zwężenie naczyń nerkowych (1,5%) które może być spowodowane dysplazją włóknisto-mięśniową i wywoływać nadciśnienie tętnicze nerkowopochodne.
Problemy poznawcze i w nauce
Najczęstszym problemem u pacjentów z NF1 jest upośledzenie poznawcze oraz w zdolności do uczenia się. Wykazano, że problemy poznawcze występują u około 80% dzieci z NF1 i mają znaczący wpływ na ich codzienne oraz szkolne życie. Najczęściej występują trudności w postrzeganiu, wykonawczym funkcjonowaniu oraz uwadze. ADHD występuje u około 38% dzieci z NF1. Występują także deficyty językowe, matematyczne i ruchowe. Wykazano, że problemy poznawcze utrzymują się na stałym poziomie do dorosłości i nie ulegają pogorszeniu, jak niektóre inne objawy fizykalne w NF-1.
Rozpoznanie
Rozpoznanie nerwiakowłókniakowatości typu 1 opiera się obecnie na kryteriach National Institutes of Health. Zgodnie z ustaleniami NIH NF-1 rozpoznać można, gdy spełnione są przynajmniej 2 z 7 warunków:
- Co najmniej 5 plam café-au-lait o średnicy >=5 mm przed okresem dojrzewania lub 6 plam o średnicy >=15 mm w późniejszym okresie
- Dwa lub więcej nerwiakowłókniaki dowolnego typu lub jeden nerwiak splotowaty (neurofibroma plexiforme)
- Piegi i (lub) przebarwienia w nieodsłoniętych okolicach ciała (okolice pachowe, pachwinowe)
- Glejak nerwu wzrokowego
- Dwa lub więcej guzki Lischa
- Charakterystyczne objawy kostne (dysplazja skrzydeł większych kości klinowej lub scieńczenie istoty zbitej trzonów kości długich z lub bez utworzenia stawów rzekomych – pseudoarthrosis)
- Krewny I° spełniający powyższe kryteria.
Do postawienia rozpoznania nie jest konieczne przeprowadzenie badań molekularnych.
Różnicowanie
Diagnostyka różnicowa nerwiakowłókniakowatości typu 1 obejmuje następujące jednostki chorobowe:
- rzadkie postaci mozaikowej lub segmentalnej nerwiakowłókniakowatości typu 1
- nerwiakowłókniakowatość typu 2 (obustronne schwannoma nerwu przedsionkowego; schwannoma nerwów czaszkowych, rdzeniowych i obwodowych; oponiaki ośrodkowego układu nerwowego; glejaki i zaćma)
- schwannomatosis (mnogie schwannomata nerwów rdzeniowych i obwodowych oraz skóry)
- zespół Noonan (ptoza, hiperteloryzm, skośne ustawienie szpar powiekowych, zrotowane do tyłu małżowiny uszne)
- fenotyp Watsona (OMIM#193520, upośledzenie umysłowe, stenoza zastawki pnia płucnego, plamy café au lait)
- plamy café au lait dziedziczone autosomalnie dominująco (OMIM114030)
- zespół McCune-Albrighta (nieregularne plamy café au lait, dysplazja włóknista kości)
- zespół LEOPARD (liczne plamy soczewicowate, hiperteloryzm, głuchota, wrodzone wady serca)
- zespół Klippla i Trénaunaya (naczyniaki skóry, przerost połowiczy, żylakowate poszerzenia żył)
- zespół Proteusza (wyrośla kostne, przerost połowiczy, hamartomata)
- mnogie tłuszczaki
- zespół Bannayana, Rileya i Ruvalcaby (mnogie tłuszczaki, naczyniaki, makrocefalia, plamy na prąciu)
- fibromatoza
- Mnoga gruczolakowatość wewnątrzwydzielnicza typu 2B
- homozygoty względem jednego z genów odpowiedzialnych za HNPCC
Leczenie
NF1 jest chorobą nieuleczalną, możliwe jest jedynie leczenie objawowe. Powinno ono być zindywidualizowane ze względu na dużą zmienność objawów. W zależności od ośrodka klinicznego preferuje się chemio- lub radioterapię w leczeniu glejaków mózgu. Kontrowersyjne jest stosowanie ketotifenu w dawkach dziennych 2–4 mg w nerwiakowłókniakach; lek miałby według niektórych zmniejszać tempo przyrostu guzów, wywierając również korzystne działanie przeciwświądowe, uspokajające i synergistyczne z lekami przeciwpadaczkowymi. Zaleca się ostrożność w leczeniu chirurgicznym szpecących zmian skórnych, ze względu na często ich nadspodziewane duże ukrwienie i nierzadko złe gojenie się ran.
W kwietniu 2020, amerykańska agencja ds. leków, FDA zatwierdziła do stosowania lek doustny selumetinib (nazwa handlowa Koselugo). Lek jest wykorzystywany u dzieci powyżej 2 roku życia.
Rokowanie
Rokowanie zależy od stopnia nasilenia zmian narządowych i właściwego prowadzenia pacjenta. Główną przyczyną przedwczesnej śmierci chorych z NF1 są choroby układu krążenia.
Przypadek Josepha Merricka
W 1909 roku wysunięto hipotezę, że słynny "Człowiek słoń", czyli Joseph Merrick (1862–1890), opisany przez sir Fredericka Trevesa w 1884 roku, cierpiał na szczególnie ciężką postać nerwiakowłókniakowatości. Nie ma jednak informacji o charakterystycznych dla choroby objawach: plamach cafe au lait czy nerwiakowłókniakach, wiadomo natomiast że Merrick miał makrocefalię, wyrośla kostne czaszki, hipertrofię kości długich, hiperplazję skóry podeszew i liczne guzy tkanek miękkich, w tym tłuszczaki. Pozwoliło to wysunąć w 1986 roku hipotezę, że Merrick miał inną, znacznie rzadszą chorobę, określaną jako zespół Proteusza. W wyniku badań genetycznych przeprowadzonych w 2003 r. przez doktora Charisa Enga ww. hipoteza wydaje się być wątpliwa.
Linki zewnętrzne
- Neurofibromatosis, type I; NF1 w bazie Online Mendelian Inheritance in Man (ang.)
- J M Friedman: Neurofibromatosis 1. GeneReviews.