
| myelosis leukaemica acuta | |
 Ostra białaczka szpikowa, płyn osierdziowy, barwienie esterazą nieswoistą | |
| Klasyfikacje | |
| ICD-10 | |
|---|---|
| C92.0 |
Ostra białaczka szpikowa |
| C92.3 |
Mięsak szpikowy |
| C92.4 |
Ostra białaczka promielocytowa |
| ICDO |
M9861/3 |
| DiseasesDB | |
| OMIM | |
| MedlinePlus | |
| MeSH | |
Ostra białaczka szpikowa, ostra białaczka mieloblastyczna, ostra białaczka nielimfoblastyczna (łac. myelosis leukaemica acuta), AML (od ang. acute myeloid leukemia), ANLL (od ang. acute nonlymphocytic leukemia) – grupa chorób spowodowana nowotworowym rozrostem w szpiku wczesnych komórek prekursorowych krwi. U jej podstaw leży klonalna proliferacja i nagromadzenie się w organizmie niedojrzałych morfologicznie i upośledzonych czynnościowo komórek blastycznych wywodzących się ze stransformowanej komórki hematopoetycznej. W mechanizmie powstawania choroby kluczowe są zaburzenia genetyczne zakłócające dojrzewanie i apoptozę wczesnych prekursorów mielopoezy. Zaburzenie hierarchicznej struktury krwiotworzenia powoduje niewydolność szpiku i objawy niedoboru krwinek czerwonych, płytek krwi i leukocytów, a jednocześnie często występuje nagromadzenie się dysfunkcyjnego klonu białaczkowego, którego nadmiar może przyczyniać się do zaburzeń mikrokrążenia, nacieczenia różnych narządów i zatrucia organizmu produktami rozpadu komórek blastycznych.
Ostra białaczka szpikowa stanowi około 80% ostrych białaczek u osób dorosłych, ale u dzieci jest nowotworem rzadszym od ostrej białaczki limfoblastycznej. Zapadalność na chorobę rośnie wraz z wiekiem. Najważniejszymi czynnikami ryzyka zachorowania jest ekspozycja na promieniowanie jonizujące, zespoły mielodysplastyczne, zespoły mieloproliferacyjne, kontakt z niektórymi czynnikami chemicznymi, a także leczenie niektórymi cytostatykami.
Rozpoznanie ostrej białaczki szpikowej jest stawiane na podstawie udziału komórek blastycznych albo ich równoważników we krwi lub szpiku przekraczającego 20%. Stwierdzenie typowych powtarzalnych zmian cytogenetycznych pozwala na rozpoznanie choroby niezależnie od odsetka blastów. W diagnostyce konieczne jest pobranie szpiku kostnego, który umożliwia szczegółowe badania, w tym badania genetyczne i molekularne pozwalające określić typ białaczki oraz zakwalifikować do odpowiedniej grupy ryzyka, co jest istotne dla wyboru najlepszej strategii leczniczej.
Leczenie ostrej białaczki szpikowej jest oparte na chemioterapii i jest złożone z trzech faz: indukcji remisji, konsolidacji (utrwalania) remisji i leczenia poremisyjnego. W indukcji remisji u chorych poniżej 60. roku życia podaje się antracykliny w połączeniu z cytarabiną. Leczenie konsolidacyjne jest zróżnicowane dla różnych grup ryzyka i w grupie korzystnego ryzyka stosuje się cytarabinę w wysokiej dawce, a w grupie pośredniego i wysokiego ryzyka wykonuje się przeszczepienie szpiku. Leczenie poremisyjne również jest zróżnicowane dla poszczególnych grup ryzyka. W grupie korzystnego ryzyka zaleca się kilka cykli cytarabiny w wysokiej dawce i monitorowanie choroby resztkowej. U chorych w grupie pośredniego i niekorzystnego ryzyka zalecaną strategią jest przeszczepienie szpiku. Dla chorych powyżej 60. roku życia stosuje się postępowanie o obniżonej toksyczności. Odmienne jest leczenie ostrej białaczki promielocytowej, gdzie najważniejszą rolę pełni tretynoina oraz trójtlenek arsenu. Ważna jest terapia wspomagająca, która zapobiega potencjalnie śmiertelnym powikłaniom choroby i może wydłużać przeżycie całkowite.
Rokowanie zależy przede wszystkim od grupy ryzyka cytogenetyczno-molekularnego i jest znacząco lepsze w grupach niższego rokowania, u których po osiągniętej remisji przeprowadzono allogeniczne przeszczepienie szpiku.
Objawy
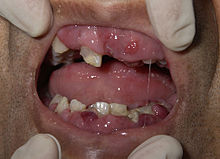

Większość objawów ostrej białaczki szpikowej jest związana z zastąpieniem prawidłowych komórek krwi komórkami nowotworowymi. W wyniku wyparcia prawidłowych prekursorów i zastąpienia ich klonem nowotworowym, który nie pełni swojej funkcji, dochodzi do jednoczesnego zaburzenia funkcji trzech linii hematopoezy niedoborów dojrzałych krwinek czerwonych, płytek krwi i funkcjonalnych krwinek białych obecnych we krwi obwodowej.
W wyniku niedoboru erytrocytów pojawiają się typowe objawy anemii, występuje osłabienie, szybka męczliwość, duszność, bladość skóry i błon śluzowych oraz przyspieszenie akcji serca. W wyniku niedoboru trombocytów rozwija się skaza krwotoczna. Małopłytkowość powoduje krwawienia z dziąseł i nosa oraz plamicę na skórze i błonach śluzowych. Może pojawić się krwawienie z przewodu pokarmowego, krwawienie z dróg rodnych i krwawienie do ośrodkowego układu nerwowego, które jest zagrożeniem dla życia chorego. Silne krwawienie może być skutkiem zespołu rozsianego wykrzepiania wewnątrznaczyniowego (DIC), który może towarzyszyć białaczce promielocytowej (APL).
Brak prawidłowych krwinek białych przyczynia się do wzmożonej podatności chorego na infekcje, mimo że we krwi często są obecne bardzo liczne prekursory dojrzałych leukocytów, które jednak są niezdolne do pełnienia funkcji fizjologicznych. Upośledzenie odporności skutkuje licznymi infekcjami, zwykle bakteryjnymi lub grzybiczymi. Występują ciężkie anginy, zapalenia płuc, zakażenia okolic odbytu oraz zmiany w jamie ustnej obejmujące bolesne afty i owrzodzenia, uaktywnienie opryszczki oraz zmiany okołozębowe. Przebieg tych zakażeń, zwłaszcza w zaawansowanym etapie choroby, może być bardzo ciężki. Jednocześnie nadmierna liczba niedojrzałych leukocytów we krwi może powodować leukostazę, czyli zaburzenia mikrokrążenia krwi spowodowane nadmierną liczbą białych krwinek. Leukostaza występuje u 5% chorych, objawia się zaburzeniami świadomości, zaburzeniami widzenia, bólem głowy oraz hipoksemią (spowodowaną upośledzeniem przepływu krwi przez płuca).
Powiększenie śledziony może występować w ostrej białaczce szpikowej, ale zwykle jest bezobjawowe. Powiększenie węzłów chłonnych występuje u 30% chorych i jest znacznie rzadsze niż w ostrej białaczce limfoblastycznej.
Często w skórze występują płaskie wykwity, które są naciekiem komórek nowotworowych. Podobne mogą pojawiać się na dziąsłach, co jest typowym objawem białaczki z linii monocytów. Nacieki mogą występować również w innych narządach, w tym układzie oddechowym oraz w sercu, co może spowodować niewydolność oddechową lub niewydolność serca. U większości chorych występują bezobjawowe nacieki w układzie moczowym. W białaczce monocytowej często dochodzi do nacieczenia opon mózgowo-rdzeniowych, w pozostałych typach jest rzadsze niż ostrej białaczce limfoblastycznej.
Rzadko pierwszym objawem jest wystąpienie litej masy pozaszpikowej zbudowanej z komórek nowotworowych, zwanej mięsakiem mieloidalnym (chloroma, pozaszpikowy guz mieloidalny). Jest to guz zbudowany z blastów, najczęściej występuje w skórze, węzłach chłonnych, kościach, przewodzie pokarmowym, jądrze i tkankach miękkich.
W przebiegu ostrej białaczki szpikowej może wystąpić zespół Sweeta, który jest paranowotworowym zapaleniem skóry.
Czasami choroba nie daje jakichkolwiek objawów i jest wykrywana przypadkowo podczas rutynowych badań krwi.
Czynniki ryzyka
Do czynników ryzyka ostrej białaczki szpikowej należą:
- wysoka dawka promieniowania jonizującego,
- czynniki chemiczne, w tym benzen, pestycydy i herbicydy,
- zespoły mielodysplastyczne, zespoły mieloproliferacyjne,
- chemioterapia za pomocą leków alkilujących i inhibitorów topoizomerazy II, szczególnie w połączeniu z radioterapią.
Zespoły mielodysplastyczne (MDS)
Zespoły mielodysplastyczne są to klonalne zaburzenia układu krwiotwórczego z obecnością nieefektywnej hematopoezy, cytopenii (zmniejszenia ilości poszczególnych składników krwi) we krwi obwodowej oraz dysplazją (nieprawidłowym ukształtowaniem) dwóch lub więcej linii krwiotworzenia, prowadzące do zwiększonego ryzyka ostrej białaczki szpikowej. Ostra białaczka szpikowa może pojawić się u 20–25% chorych z zespołem mielodysplastycznym. Przebieg choroby oraz ryzyko rozwinięcia ostrej białaczki szpikowej zależy od udziału liczby blastów w szpiku kostnym, rodzaju nieprawidłowości chromosomowych oraz nasilenia cytopenii. Rokowanie zależy również od wieku chorego, płci i konieczności transfuzji. Ryzyko progresji jest znacznie większe w grupach z gorszym rokowaniem.
| Czynniki rokowniczy | Liczba punktów | ||||||
| 0 | 0,5 | 1 | 1,5 | 2 | 3 | 4 | |
| Blasty w szpiku (%) | ≤2 | – | >2–<5 | – | ≥5–≤10 | >10 | – |
| Zmiany cytogenetyczne | bardzo korzystne | – | korzystne | – | pośrednie | niekorzystne | bardzo niekorzystne |
| Hemoglobina | ≥10 | – | ≥8–<10 | <8 | – | – | – |
| Płytki (w tys.) | ≥100 | ≥50–100 | <50 | – | – | – | – |
| Liczba neutrofili | ≥0,8 | ≤0,8 | – | – | – | – | – |
| Kategorie ryzyka IPSS-R | |||||||
| bardzo niskie | niskie | pośrednie | wysokie | bardzo wysokie | |||
| Grupy ryzyka | 1,5–3 | >1,5–3 | >3–4,5 | >4,5–6 | >6 | ||
| Mediana przeżycia | 5,4 | 4,8 | 2,7 | 1,5 | 0,7 | ||
| Mediana progresji do AML u 25% z MDS (lata) | ? | 9,4 | 2,5 | 1,7 | 0,7 |
Czynniki chemiczne
Zawodowy kontakt z benzenem zwiększa ryzyko zachorowania na ostrą białaczkę szpikową. Występuje związek pomiędzy wielkością narażenia na benzen a ryzykiem powstawania ostrej białaczki szpikowej. Palenie tytoniu zwiększa ryzyko zachorowania na ostrą białaczkę szpikową. Pestycydy i herbicydy również należą do czynników ryzyka zachorowania.
Białaczka wtórna do chemioterapii
Część przypadków ostrej białaczki szpikowej jest związana z leczeniem przeciwnowotworowym, z którym największym ryzykiem obarczona jest chemioterapia za pomocą leków alkilujących i inhibitorami topoizomerazy (etopozyd, tenipozyd). Jest to najczęściej rozpoznawany wtórny nowotwór po chemioterapii. Białaczki wtórne stanowią niejednolitą grupę chorób zdefiniowaną cytogenetycznie. Białaczki powstałe po leczeniu lekami alkilującymi zwykle wykazują nieprawidłowości w chromosomach 5 lub 7 (głównie delecje), a w białaczkach wtórnych do inhibitorów topoizomerazy II zamianami w 11q. Znacznie rzadziej w związku z leczeniem mogą powstawać typy z mutacjami t(15;17), inv(16) i t(8;21). Zwiększone ryzyko występuje w okresie od 2 do 10 lat po zakończonym leczeniu. Inhibitory topoizomerazy charakteryzują się krótszym okresem latencji (okresu od zakończenia leczenia tymi lekami do wystąpienia objawów choroby) w stosunku do leków alkilujących. Skumulowane 10-letnie ryzyko wystąpienia ostrej białaczki szpikowej po leczeniu raka piersi wynosi 0,7%, a po leczeniu chłoniaka Hodgkina wynosi 2–10%, choć te w przypadku chłoniaka Hodgkina jest związane z radioterapią. Część badań wskazuje, że po 10 latach od zakończenia leczenia nie ma zwiększonego ryzyka zachorowania, jednak inne nie potwierdzają tego. Różnice mogą wynikać z metodologii tych badań.
Promieniowanie jonizujące
Narażenie na promieniowanie jonizujące jest czynnikiem ryzyka wystąpienia ostrej białaczki szpikowej. Wielkość ryzyka prawdopodobnie jest proporcjonalna do otrzymanej dawki promieniowania.
Czynniki genetyczne
Ważnym czynnikiem genetycznym jest zespół Downa, chorzy mają 10-20-krotnie zwiększone ryzyko zachorowania na ostrą białaczkę szpikową w porównaniu do osób zdrowych, choć ryzyko rozwinięcia niektórych typów jest znacznie większe. Ostra białaczka megakarioblastyczna występuje 500-krotnie częściej niż u chorych bez zespołu Downa.
Niedokrwistość Fanconiego, zespół Blooma, zespół ataksja-teleangiektazja, zespół Shwachmana-Diamonda, zespół Noonan, choroba Kostmanna, dyskeratoza wrodzona (dyskeratosis congenita), niektóre wrodzone choroby trombocytów są czynnikami ryzyka zachorowania na ostrą białaczkę szpikową.
Rozpoznanie

Rozpoznanie ostrej białaczki szpikowej wymaga pobrania krwi obwodowej i szpiku kostnego oraz wykonania dalszych szczegółowych badań. Podstawą do rozpoznania choroby jest stwierdzenie odsetka ≥20% blastów (mieloblastów lub ich równoważników: monoblastów, promonocytów, megakarioblastów) w szpiku lub we krwi obwodowej. Blasty są to wczesne niedojrzałe komórki prekursorowe, które zwykle wykazują ekspresję pewnych wspólnych markerów linii mieloidalnej (CD13, CD33) i jednocześnie markerów komórek macierzystych szpiku (CD34, rzadziej CD117).
Obecność pewnych powtarzalnych zmian cytogenetycznych, w tym t(8;21), t(15;17), inv (16), t(16;16), pozwala rozpoznać ostrą białaczkę szpikową bez względu na odsetek blastów. Erytroblasty nie są liczone jako blasty z wyjątkiem ostrej białaczki erytroblastycznej. W przypadku wartości odsetka blastów w szpiku kostnym w zakresie 6–19% umownie rozpoznaje się zespół mielodysplastyczny. Mięsak mieloidalny jest rozpoznawany na podstawie obrazu morfologicznego szpiku. Szczegółowe rozpoznanie typu AML opiera się na badaniach cytogenetycznych, molekularnych i immunofenotypowych.
Badanie krwi obwodowej
Badanie morfologii krwi jest badaniem wstępnym, jednak może wykazać nieprawidłowości ostatecznie nakierowujące na rozpoznanie ostrej białaczki szpikowej. Wyparcie pozostałych linii hematopoetycznych ze szpiku i zastąpienie ich klonem nowotworowym znajduje odzwierciedlenie w obrazie krwi obwodowej. W morfologii zwykle występuje umiarkowana leukocytoza, ale możliwa jest leukopenia. Leukocytoza powyżej 100 000/μl występuje tylko u 5% chorych. W rozmazie występują komórki blastyczne, jednak ich odsetek jest różny w zależności od typu i etapu choroby. Stwierdzenie >1000–2000 blastów/μl wystarcza do postawienia diagnozy ostrej białaczki szpikowej, przez co pobranie szpiku i szczegółowe badania nie opóźniają rozpoczęcia leczenia.
W obrazie choroby obserwuje się wyparcie dojrzałych komórek. U większości chorych w morfologii krwi stwierdza się znaczną niedokrwistość i małopłytkowość, a liczba neutrofilów nie przekracza 1000/μl. Niedokrwistość jest normochroniczna (prawidłowy poziom MCH) i normocytowa (prawidłowy poziom MCV).
Możliwe jest występowanie tzw. przerwy białaczkowej, czyli brak lub występowanie tylko nielicznych form pośrednich obok bardzo licznych komórek blastycznych i nielicznych komórek dojrzałych.
Szpik kostny
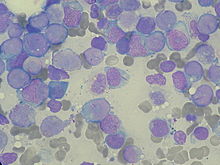
Przed rozpoczęciem leczenia należy pobrać szpik kostny. Rutynowo jest wykonywana biopsja aspiracyjna szpiku, a w niektórych przypadkach konieczna jest trepanobiopsja. Ocena szpiku pozwala ustalić wstępne rozpoznanie, rozpoznać postać choroby bez specyficznych zmian cytogenetycznych oraz ocenić komórkowość szpiku, zmiany zrębowe i wzór dojrzewania. Zaleca się ocenę przynajmniej 500 komórek z rozmazu szpiku. Pobranie szpiku dostarcza materiału do badań cytogenetycznych i molekularnych, które pozwalają ustalić typ białaczki oraz czynniki rokownicze. Szpik po wybarwieniu metodą Maya-Grünwalda-Giemsy lub Wrighta-Giemsy jest badany pod mikroskopem optycznym, a następnie poddawany immunofenotypowaniu metodą cytometrii przepływowej oraz badaniom genetycznym. Ocena liczby blastów za pomocą oznaczenia CD34 w cytometrii przepływowej nie jest zalecana jako substytut oceny wzrokowej. Morfologia komórek nowotworowych jest różna w poszczególnych typach. Jądro blastu zawiera niespecyficzne ziarnistości, jego chromatyna jest rozproszona, występuje jedno lub więcej jąderek. Nieprawidłowe ziarnistości azurofilne układają się w kształt fioletowoczerowonych pałeczek i są nazywane pałeczkami Auera. Ich obecność jednoznacznie wskazuje na pochodzenie mieloidalne blastów.
Badania cytochemiczne
Badania cytochemiczne pomagają odróżnić ostrą białaczkę szpikową od ostrej białaczki limfoblastycznej, a także często określić typ morfologiczny AML. Metoda wymaga pobrania szpiku kostnego. Największe znaczenie z badań cytochemicznych ma oznaczenie aktywności mieloperoksydazy (MPO/POX), aktywności esterazy nieswoistej (NSE), obecności lipidów za pomocą Sudanu B (SB) oraz oznaczenie obecności glikogenu (odczynnik Schiffa, reakcja PAS). Stwierdzenie mieloperoksydazy u więcej niż 3% blastów wskazuje na białaczkę zróżnicowaną, ale jej brak nie wyklucza linii mieloidalnej. Podobne zastosowanie ma Sudan B, choć jest mniej specyficzny. Oznaczenie aktywności esterazy nieswoistej wykazuje jej rozproszoną aktywność w cytoplazmie monoblastów (w 80% przypadków reakcja pozytywna) i monocytów (w 20% przypadków reakcja pozytywna). Reakcja PAS daje pozytywny wynik w ostrej białaczce erytroblastycznej (erytroleukemia). Barwienia cytoenzymatyczne w coraz większym stopniu są zastępowane badaniem immunofenotypowym za pomocą cytometrii przepływowej. Badania cytochemiczne w znacznym stopniu zostały zastąpione przez inne badania i obecnie nie są wymagane do diagnozy.
Immunofenotypowanie
Immunofenotypowanie jest to oznaczenie występowania pewnych antygenów (immunofenotyp) za pomocą metod immunochemicznych lub cytometrycznych. W diagnostyce ostrej białaczki szpikowej stosuje się cytometrię przepływową przy zastosowaniu co najmniej 3–4 parametrów. Nie ma konsensusu co do wysokości punktu odcięcia przy jakim uważa się dany marker za pozytywny. Dla większości markerów przyjęto, że przynajmniej 20% komórek białaczkowych musi wykazywać ekspresję tego markera, ale dla niektórych markerów dolna granica została zdefiniowana przy 10%. Immunofenotypowanie jest niezbędne do ustalenia rozpoznania ostrej białaczki szpikowej mało zróżnicowanej, ostrej białaczki megakarioblastycznej i ostrej białaczki o niejednoznacznym pochodzeniu liniowym.
Ostrą białaczkę szpikową mało zróżnicowaną cechuje brak morfologicznych i cytochemicznych cech dojrzewania, a w większości przypadków są obecne markery wczesnej hematopoezy (CD34, CD38 i HLA-DR) i brak większości antygenów świadczących o dojrzewaniu. Mieloperoksydaza jest niewykrywalna metodami cytochemicznymi, ale w cytometrii przepływowej może być rozpoznana.
Ostra białaczka megakarioblastyczna jest białaczką, w której w szpiku występuje co najmniej 20% blastów i 50% z nich stanowi linię megakariocytową, a megakarioblasty typowo posiadają markery CD41 i/lub CD61, a rzadziej CD42.
Ostre białaczki szpikowe o niejednoznacznym pochodzeniu liniowym obejmują przypadki braku zróżnicowania liniowego (ostra białaczka niezróżnicowana – acute undifferentiated leukemia (AUL)) oraz takie, w których blasty zawierają markery więcej niż jednej linii (ostra białaczka o mieszanym fenotypie – mixed phenotype acute leukemia (MPAL)). Ostra białaczka niezróżnicowana często wykazuje ekspresje HLA-DR, CD34 lub CD38 i nie ma markerów liniowych. Ostra białaczka o mieszanym fenotypie może zawierać odrębne populacje z różnych linii lub jedną populację z markerami różnych linii albo jest kombinacją tych możliwości. Cytometria przepływowa w oparciu o antygeny stwierdzone w danym klonie białaczkowym jest wykorzystywana do oceny występowania minimalnej choroby resztkowej.
| Linie | Markery |
| Markery prekursorowe | CD34, CD38, CD117, CD133, HLA-DR |
| Markery granulocytowe | CD13, CD15, CD16, CD33, CD65, MPO w cytoplazmie |
| Markery monocytowe | NSE, CD11c, CD14, CD64, lizozym, CD4, CD11b, CD36 |
| Markery megakariocytowe | CD41, CD61, CD42 |
| Markery erytroidalne | CD235a |
Badania cytogenetyczne i diagnostyka molekularna
Badania cytogenetyczne są standardowym badaniem chorego z podejrzeniem ostrej białaczki szpikowej. Pewne zaburzenia cytogenetyczne są często obserwowane w ostrej białaczce szpikowej, a część z nich jest związana z poszczególnymi typami morfologicznymi ostrej białaczki szpikowej. Nieprawidłowości chromosomalne są stwierdzane u ponad 50% chorych z ostrą białaczką szpikową. Obecna klasyfikacja choroby według WHO w znacznej mierze opiera się na stwierdzeniu charakterystycznych, powtarzalnych zmian cytogenetycznych i stwierdzenie niektórych z nich upoważnia do rozpoznania choroby niezależnie od liczby blastów w szpiku czy krwi obwodowej. Stwierdzenie poszczególnych zmian genetycznych ma podstawowe znaczenie rokownicze i przekłada się na strategię leczniczą dopasowaną do grup ryzyka. Do oceny wymagana jest biopsja szpiku i ocena przynajmniej 20 komórek w metafazie. Komórki z nieprawidłowym kariotypem również mogą być ocenione z krwi obwodowej. Powtarzalne aberracje chromosomowe są wykrywane za pomocą klasycznego badania cytogenetycznego i uzupełniająco fluorescencyjną hybrydyzacją in situ.
Krew obwodowa oraz szpik kostny są poddawane badaniom molekularnym. RNA i DNA są ekstrahowane, a komórki są zamrażane. Metodą łańcuchowej reakcji polimerazy z odwrotną transkrypcją ocenia się obecność niektórych genów fuzyjnych oraz mutacji somatycznych związanych z białaczką.
Badania laboratoryjne
W wyniku samoistnego lub indukowanego leczeniem rozpadu klonu nowotworowego dochodzi do uwolnienia zawartości ich komórek. U chorych na ostrą białaczkę szpikową stwierdza się podwyższone stężenie kwasu moczowego (hiperurykemia), podwyższone stężenie potasu (hiperkaliemia) oraz podwyższoną aktywność LDH. W badaniach laboratoryjnych może wystąpić rzekoma hipoglikemia i hipoksemia spowodowana bardzo wysoką leukocytozą i zużywaniem przez nie tlenu i glukozy w pobranej próbce do badania.
Klasyfikacja
Klasyfikacja WHO z 2008
Obecnie obowiązującą klasyfikacją ostrej białaczki szpikowej jest klasyfikacja WHO z 2008 roku. Jest ona oparta na cechach morfologicznych, immunofenotypowych, nieprawidłowościach molekularnych i cytogenetycznych. Ostre białaczki szpikowe z powtarzalnymi zmianami cytogenetycznymi są kwalifikowane do grupy na podstawie stwierdzonych nieprawidłowości cytogenetycznych.
Ostre białaczki szpikowe związane ze zmianami mielodysplastycznymi są rozpoznawane w trzech sytuacjach:
- stwierdzenie historii zespołu mielodysplastycznego lub nowotworu mielodysplastycznego/mieloproliferacyjnego, które następnie ewoluuje do AML i zawartość odsetka blastów wynosi powyżej 20%,
- co najmniej 50% komórek albo dwie linie mieloidalne są dysplastyczne,
- stwierdzenie charakterystycznych zmian cytogenetycznych: del(7q), del(5q), i(17q) lub t(17p), del(13q), del(11q), del(12p) lub t(12p), del(9q), idic(X)(q13), t(11;16)(q23;p13.3), t(3;21)(q26.2;q22.1), t(1;3)(p36.3;q21.1), t(2;11)(p21;q23), t(5;12)(q33;p12), t(5;7)(q33;q11.2), t(5;17)(q33;p13), t(5;10)(q33;q21), t(3;5)(q25;q34).
Nowotwory mieloidalne zależne od terapii są odrębną jednostką, choć ze względu na polichemioterapię nie zawsze jest możliwe dokładne ustalenie podtypu.
| Typ | Podtyp (zmiany cytogentyczne) |
| Ostre białaczki z powtarzalnymi zmianami cytogenetycznymi | |
| AML z t(8;21)(q22;q22) – RUNX1-RUNX1T1 | |
| AML z inv(16)(p13;1q22) lub t(16;16)(p13.1;q22) – CBFB-MYH11 | |
| Ostra białaczka promielocytowa t(15;17)(q22;q12) – PML-RARA | |
| AML z 11q23 – MLL, t(9;11)(p22;q23) – MLLT3-MLL | |
| AML z t(6;9)(p23;q34) – DEK-NUP214 | |
| AML z inv(3)(q21;q26.2) lub t(3;3)(q21;q26.2) – RPN1-EVI1 | |
| AML megakarioblastyczna z t(1;22)(p13;q13) – RBM15-MKL1 | |
| AML z mutacją NMP1 (jednostka prowizoryczna) | |
| AML z mutacją CEBPA (jednostka prowizoryczna) | |
| AML związane ze zmianami mielodysplastycznymi (AML z dysplazją wieloliniową) | |
| Nowotwory mieloidalne zależne od terapii (wtórna AML) | |
| AML bez specyfikacji innej niż morfologiczna | |
| Ostra białaczka szpikowa mało zróżnicowana (dawniej FAB M0) | |
| Ostra białaczka szpikowa bez cech dojrzewania (dawniej FAB M1) | |
| Ostra białaczka szpikowa z dojrzewaniem (dawniej FAB M2) | |
| Ostra białaczka szpikowa z dojrzewaniem (dawniej FAB M3) | |
| Ostra białaczka mielomonocytowa (dawniej FAB M4) | |
| Ostra białaczka monoblastyczna i monocytowa (dawniej FAB M5) | |
| Ostra białaczka erytroblastyczna (dawniej FAB M6) | |
| Ostra białaczka megakarioblastyczna (dawniej FAB M7) | |
| Ostra białaczka bazofilowa | |
| Ostra panmieloza z mielofibrozą | |
| Mięsak mieloidalny | |
| Proliferacje mieloidalne związane z zespołem Downa | Przemijająca nieprawidłowa mielopoeza |
| Białaczka szpikowa związana z zespołem Downa | |
| Ostre białaczki o niejednoznacznym pochodzeniu liniowym |
Klasyfikacja FAB
Klasyfikacja francusko-amerykańsko-brytyjska obecnie ma znaczenie historyczne, choć bywa stosowana. Została opracowana w roku 1976 i jest oparta na cechach morfologicznych i cytochemicznych.
| Typ FAB | Nazwa |
| M0 |
 Ostra białaczka szpikowa o bardzo niskim stopniu zróżnicowania Ostra białaczka szpikowa o bardzo niskim stopniu zróżnicowania
|
| M1 |
 Ostra białaczka mieloblastyczna bez cech dojrzewania Ostra białaczka mieloblastyczna bez cech dojrzewania
|
| M2 |
 Ostra białaczka mieloblastyczna z cechami dojrzewania Ostra białaczka mieloblastyczna z cechami dojrzewania
|
| M3 |
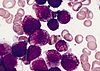 Ostra białaczka promielocytowa Ostra białaczka promielocytowa
|
| M4 |
 Ostra białaczka mielomonocytowa Ostra białaczka mielomonocytowa
|
| M5a |
 Ostra białaczka monoblastyczna (monocytowa słabo zróżnicowana) Ostra białaczka monoblastyczna (monocytowa słabo zróżnicowana)
|
| M5b |
 Ostra białaczka monocytowa (monocytowa dobrze zróżnicowana) Ostra białaczka monocytowa (monocytowa dobrze zróżnicowana)
|
| M6 |
 Erytoroleukemia Erytoroleukemia
|
| M7 |
 Ostra białaczka megakariocytowa Ostra białaczka megakariocytowa
|
Patogeneza ostrej białaczki szpikowej

Ostra białaczka szpikowa charakteryzuje się niekontrolowanym wytwarzaniem i akumulacją niedojrzałych, nieprawidłowych komórek hematopoetycznych. Jednocześnie nagromadzenie się komórek nowotworowych upośledza produkcję prawidłowych elementów morfotycznych krwi. Klon komórki nowotworowej powstaje w wyniku transformacji komórki macierzystej lub wczesnych komórek progenitorowych. U podstaw transformacji nowotworowej leżą zaburzenia genetyczne, które ostatecznie zakłócają regulację podziałów oraz różnicowanie się komórek. Ostra białaczka szpikowa może powstać de novo, jak to ma miejsce w 80% przypadków, lub po etapie przewlekłym obejmującym zespoły mielodysplastyczne i zespoły mieloproliferacyjne – jako wtórna ostra białaczka szpikowa.
Fizjologicznie hematopoeza jest zorganizowana w sposób hierarchiczny. Spoczynkowe komórki macierzyste są prekursorami kolejnych typów komórek, z których w wyniku procesu dojrzewania ostatecznie powstają dojrzałe komórki krwi. Komórki macierzyste mają zdolność do ciągłego podziału komórkowego i jednocześnie do samoodnawiania się, która zależy między innymi od telomerazy, która jest enzymem zapobiegającym skracaniu telomerów i ograniczaniu liczby możliwych podziałów. Komórki potomne stopniowo tracą zdolność do samoodnowiania się, co jest cechą komórek macierzystych, które są nieśmiertelne, jednocześnie jednak komórki potomne stają się bardziej aktywne mitotycznie. Liczba komórek macierzystych jest regulowana przez apoptozę.
W ostrej białaczce szpikowej dochodzi do stopniowego nagromadzenia zaburzeń genetycznych, stopniowo wykształcając klon wczesnych komórek prekursorowych z zaburzeniami podziałów komórkowych, zahamowaniem różnicowania, nabytą zdolnością do samoodnawiania się, zablokowaniem apoptozy, niestabilnością genetyczną i zaburzoną kontrolą cyklu komórkowego. W wyniku wzmożonych podziałów, zaburzonego różnicowania się i zmniejszonej wrażliwości na apoptozę dochodzi do gromadzenia się niedojrzałych komórek. Zmniejszenie śmierci komórek nowotworowych jest uważane za główny mechanizm powstawania tej choroby.
Zgodnie z teorią „dwóch uderzeń” dla powstania choroby kluczowe jest wystąpienie zmian genetycznych stymulacyjnych proliferację lub przeżycie oraz zmian prowadzących do zahamowania dojrzewania. Zmianami genetycznymi są zarówno mutacje cytogenetyczne lub zmiany epigenetyczne – zmiany ekspresji genów bez zmian w materiale genetycznym.
Niewydolność szpiku
W ostrej białaczce szpikowej dochodzi do niewydolności szpiku. Mimo że w szpiku znajdują się często bardzo liczne nowotworowo zmienione komórki progenitorowe, to w wyniku zaburzeń dojrzewania nie powstają z nich dojrzałe i funkcjonalne komórki, które mogłyby pojawić się we krwi. Brak dojrzałych komórek powoduje objawy niedokrwistości, zaburzeń krzepnięcia, a także upośledzenie odporności. Objawy te powodują znaczną chorobowość i śmiertelność z powodu białaczki, pogarszając jakość życia chorych. Niewydolność utrudnia leczenie, uniemożliwiając zastosowanie odpowiednio intensywnej chemioterapii.
Namnożenie dużej liczby nieulegających dojrzewaniu i akumulujących się komórek nowotworowych skutkuje wyparciem prawidłowych komórek progenitorowych. Prawdopodobnie proces ten w mniejszym stopniu jest spowodowany samym zmniejszeniem liczby prawidłowych komórek macierzystych, a raczej zahamowaniem ich różnicowania przez oddziaływanie z komórkami nowotworowymi. Proces prawdopodobnie odbywa się za pośrednictwem chemokin.
Niewydolność szpiku wydaje się być procesem odwracalnym.
Mutacje genetyczne i epigenetyczne
W ostrej białaczce szpikowej nielosowe aberracje chromosomowe są obserwowane u 50–60% chorych. Tylko niektóre mutacje dają korzyści selektywne i mają znaczenie w patogenezie (tzw. „mutacje kierujące”) i rokowaniu choroby, w odróżnieniu od „mutacji tła”, które nie dają przewagi selektywnej. Nieprzypadkowe zmiany genetyczne obejmują translokacje zrównoważone, w tym t(8;21)(q22;q22) i t(15;17)(q22;q21), delecje (del(5q), del(7q)) oraz inwersje (inv(3), inv(8), inv(16)), trisomie, liczne mutacje punktowe oraz zmiany epigenetyczne. Wiele z odkrytych mutacji wpływa na te same mechanizmy powodujące chorobę, wywierając podobny skutek, a także istnieje wiele różnych możliwości mutacji pojedynczego genu. Dodatkowo niektóre geny podlegają zmianom niestrukturalnym (zmiany epigenetyczne), które mogą je wyłączać i wpływać na powstawanie choroby.
Prawdopodobnie do wywołania ostrej białaczki szpikowej konieczne jest powstanie więcej niż jednej mutacji genetycznej (teoria dwóch uderzeń). W wielostopniowej patogenezie ostrej białaczki szpikowej dochodzi do zmian w obrębie dwóch grup komplementarnych mutacji. Do pierwszej grupy zalicza się geny odpowiadające za proliferację i zwiększające przeżycie komórek progenitorowych. Są to geny FLT3, RAS i KIT, rzadziej BCL/ABL i TEL/PDGFβR. Choć mutacje genów tej grupy występują u około 50% chorych, to rzadko są obserwowane razem u tego samego chorego. Do drugiej grupy zalicza się geny odpowiadające za różnicowanie i samoodnawianie (unieśmiertelnienie). Należą do niej CBF, PML/RARα, mutacje MLL, mutacje HOX, zmiany w obrębie koaktywatorów p300/TIF2 i CBP. Również mutacje tej grupy niemal nigdy nie występują razem u tego samego chorego. Mutacje tej grupy, powodując zaburzenie różnicowania i unieśmiertelniając klon, jako samodzielne mutacje nie umożliwiają powstania ostrej białaczki szpikowej. Z kolei jednoczesne mutacje genów z obu grup bardzo często występują łącznie. Do kolejnych mutacji dochodzi w wyniku nasilenia proliferacji i narastającej niestabilności genetycznej. Prowadzi to do oligoklonalności nowotworu, gdzie występuje kilka populacji o różnych posiadanych mutacjach, a wczesne mutacje dotyczą większości komórek białaczkowych. Faktycznie model dwóch uderzeń wydaje się modelem uproszczonym, ponieważ już w etapie przewlekłym AML można obserwować kilka mutacji i mutacje obu klas są częścią bardziej złożonego obrazu, szczególnie wobec badań podkreślających znaczenie zmian epigenetycznych. Dodatkowo niektóre badania sugerują, że mutacje muszą zachodzić w odpowiednim etapie rozwoju białaczki w określonej kolejności.
Niektóre mutacje są związane z kliniczno-fenotypowym typem białaczki. Translokacja genu MLL zlokalizowanego w 11q23 jest związana z fenotypem mielomonocytowym lub monocytowym (dawniej FAB M4 i M5), z kolei t(15;17)(q22;q21) jest związana z fenotypem promielocytowym (FAB M3). Występowanie pewnych powtarzalnych mutacji jest podstawą do klasyfikacji AML.
Mutacje, szczególnie translokacje, mogą powodować powstawanie genów fuzyjnych, które zawierają sekwencje obu genów macierzystych. W konsekwencji występowania genów fuzyjnych powstają białka fuzyjne o zmienionych właściwościach biologicznych.
Translokacje chromosomalne modyfikujące Core-Binding Factor

Core-Binding Factor (CBF) jest czynnikiem transkrypcyjnym odpowiedzialnym za różnicowanie się komórek. Czynnik jest heterodimerem i składa się z 3 różnych podjednostek CBFa – RUNX1, RUNX2 i RUNX3 oraz wspólnej podjednostki CBFb niewiążącej DNA. Czynnik jest niezbędny do wytworzenia komórek macierzystych szpiku i wystąpienia hematopoezy. Homozygotyczny brak obu alleli genu RUNX1 lub CBFB uniemożliwia hematopoezę i powoduje śmierć zarodka. Odpowiada za transkrypcję wielu genów ważnych dla rozwoju białaczki, w tym za geny odpowiadające za produkcję cytokin, również GM-CSF, oraz za receptory cytokin.
Białaczki ze zmianami CBF należą do najczęstszych podtypów cytogenetycznych, zmiany są wykrywane w około 13–15% przypadków AML. Translokacja t(8;21) powoduje przeniesienie genu RUNX1 z 21q22 do genu RUNX1T1 (ETO) z 8q22, powoduje to powstanie genu fuzyjnego RUNX1/RUNX1T1 (AML1-ETO). Z kolei inwersja w chromosomie 16 i translokacja t(16;16) powodują przeniesienie genu CBFB z genem miozyny mięśni gładkich (MYH11) zlokalizowanego 16q22, powodując powstanie genu fuzyjnego CBFB/MYH11.
Translokacja t(8;21) występuje u młodszych chorych w 5% przypadków ostrej białaczki szpikowej, a inv(16) lub t(16;16) jest znajdowana w 5–8% przypadków tej choroby. Translokacja t (8;21) jest często obecna w typie morfologicznym FAB M2 (ostra białaczka mielobastyczna z cechami dojrzewania), występując w 40% jej przypadków, a inv(16) z FAB M4Eo (ostra białaczka szpikowa mielomonocytowa z eozynofilią). Rzadziej inv(16) występuje z podtypie FAB M2, M5 i M4.
W konsekwencji powstania białek fuzyjnych dochodzi do aktywacji kolejnych czynników transkrypcyjnych i białek regulatorowych, które ogrywają rolę w powstawaniu i progresji ostrej białaczki szpikowej, a także zaburzając różnicowanie komórek nowotworowych. Choć białka fuzyjne RUNX1-RUNX1T1 i CBFB-MYH11 są krytyczne do wystąpienia białaczki, to samodzielne ich występowanie nie jest wystarczające do wystąpienia choroby i konieczne są inne zmiany genetyczne lub epigenetyczne.
Mutacja CBF należy do korzystnych czynników ryzyka.
Translokacje chromosomalne modyfikujące receptor alfa kwasu retynoinowego
W 95% ostrej białaczki promielocytowej (podtyp FAB M3) wykrywa się translokację t(15;17)(q22;q21). Translokacja powoduje połączenie genu białka białaczki promielocytowej (PML) będącym czynnikiem transkrypcyjnym (palcem cynkowym) i genem supresorowym, z receptorem α kwasu retynoinowego (RARα). Powoduje to powstanie białka fuzyjnego PML/RARα, który hamuje transkrypcję i blokuje różnicowanie komórkowe, ograniczając odpowiedź na kwas retinowy. Dochodzi do zatrzymania różnicowania w fazie promielocyta. Dodatkowo białko fuzyjne zakłóca działanie białka PML, które działa jako antyonkogen. Leczenie kwasem retinowym powoduje związanie białka fuzyjnego PML/RARα i odwrócenia tłumienia genów docelowych niezbędnych do prawidłowego rozwoju układu krwiotwórczego.
Translokacje chromosomalne w genach HOX
Geny HOX pełnią wiele ważnych funkcji podczas rozwoju zarodkowego kręgowców. Funkcja tych genów jest kluczowa dla prawidłowej proliferacji i różnicowania się macierzystych i progenitorowych komórek macierzystych. W ostrej białaczce szpikowej często dochodzi do nadekspresji tych genów. Geny HOX ulegają bardzo ścisłej ekspresji w kolejności zgodnej z ich ułożeniem w chromosomie (kolinearność). Prawidłowo gen HOXA9 ulega ekspresji we wczesnym etapie hematopoezy, a w dojrzałych komórkach jego ekspresja nie jest wykrywalna.
Geny HOX mogą być rozregulowane w wyniku kilku różnych mechanizmów. Może do nich dochodzić w wyniku translokacji chromosomalnej. Translokacja t(7;11) powoduje fuzję genów NUP98 i HOXA9, a translokacja t(2;11) fuzję genów NUP98 z HOXD13. Translokacje t(v;11q23) powodują rearanżacje genu MLL (mixed-lineage leukemia) i zaburzeń funkcji genów HOX, wówczas często obserwuje się nadekspresję HOXA4, FOXA9, HOXA10, PBX, MEIS1 (myeloid ecotropic viral integration site 1). Translokacja t(8;16)(p11;p13) prowadzi do powstawania białka fuzyjnego MYST3-CREBBP, które również jest związane z nadekspresją HOXA9, HOXB9, HOXA10 i MEIS1.
Prawdopodobnie nieprawidłowo regulowana i nadmierna ekspresja HOXA9 z aktywnym promotorem NUP98 powoduje nieprawidłowe różnicowanie. NUP98 może rekrutować niektóre aktywatory transkrypcyjne, w tym CBP/p300. W modelach zwierzęcych sama nadekpresja HOXA9 jest niewystarczająca do złośliwej transformacji, jednak dochodzi do niej w obecności aktywatorów transkrypcji. Nadekspresja HOXA9 i Meis1, którą mogą powodować białka fuzyjne MLL, są wystarczające dla przemiany białaczkowej szpiku kostnego u myszy.
W 90% przypadków ostrej białaczki szpikowej nadekspresji ulega czynnik transkrypcyjny CDX2 (caudal-type homeobox transcription factor 2), który jest związany z określeniem osi przód-tył podczas rozwoju embrionalnego oraz różnicowaniem się komórek nabłonka jelitowego, regulując ekspresję genów HOX. W warunkach fizjologicznych nie ulega on ekspresji w komórkach progenitorowych szpiku kostnego. Prawdopodobnie wysoki poziom ekspresji CDX2 powoduje upośledzenie różnicowania się komórek progenitorowych. W badaniach na myszach wykazano, że nadmierna ekspresja CDX2 powoduje ostrą białaczkę szpikową oraz nadmiermierną ekspresję HOXB6. Zaburzona ekspresja CDX2 jest stwierdzana w 25% AML, co może wskazywać, że CDX2 jest ważną drogą prowadzącą do ostrej białaczki szpikowej.
Mutacje chromosomalne w obrębie genu MLL

Zrównoważone translokacje chromosomalne obejmujące gen MLL (mixed-lineage leukemia) powiązano w niejednorodną grupę białaczek, obejmujące ostrą białaczkę szpikową, ostrą białaczkę limfoblastyczną, białaczki o mieszanym fenotypie i białaczki wtórnej do leczenia inhibitorami topoizomerazy II. Gen jest zlokalizowany na długim ramieniu chromosomu 11 (11q23).
Mutacje 11q23, które zakłócają funkcję genu MLL obejmują 6% AML i 5–10% ALL. Jednak w szczególnych grupach chorych odsetek ten jest większy, u dzieci poniżej 12 miesiąca życia zmiany genetyczne genu MLL występują u 80% chorych niemowląt. Również u chorych leczonych inhibitorami topoizomerazy II częściej dochodzi do rearanżacji tego genu.
Gen MLL w ostrej białaczce szpikowej i limfoblastycznej często jest objęty różnymi translokacjami, których opisano około 40–50. Mimo dużej liczby opisanych kombinacji tylko 5 translokacji stanowi 80% wszystkich występujących w ostrej białaczce szpikowej: t(4;11)(q21;q23) – MLL-AF4, t(6;11)(q27;q23) – MLL-AF6, t(9;11)(p22;q23) – MLL-AF9, t(11;19)(q23;p13.1) – MLL-ELL, t(11;19)(q23;p13.3) – MLL-ENL. W większości gen jest związany z ostrą białaczką mielomonocytową (FAB M4) oraz monoblastyczną i monocytową (FAB M5), a rzadziej z ostrą białaczką szpikową z dojrzewaniem (FAB M2).
Geny fuzyjne MLL mogą wywołać AML, jednak mechanizm skutkujący tym następstwem nie jest poznany. Przypuszcza się, że białko fuzyjne MLL może blokować różnicowanie komórek hematopoetycznych oraz zwiększać ich apoptozę. Białko MLL powszechnie ulega ekspresji podczas rozwoju oraz później w wielu tkankach, w tym również komórkach mieloidalnych i limfoidalnych. MLL wiąże się do regionów regulatorowych genów HOX, w tym HOXA9 i HOXC8. Ekspresja białek fuzyjnych MLL prowadzi do nadekspresji genów HOXA7, HOXA9 i Meis1, co prawdopodobnie tłumaczy onkogenność tego transkryptu. Nadekspresja HOXA9 i Meis1 są wystarczające dla przemiany białaczkowej szpiku kostnego u myszy. Jednak nadekspresja HOXA9 nie jest wymagana do wywołania białaczki w fuzji MLL-GAS7, gdzie występują niedobory HOXA9. Możliwe, że zwiększenie ekspresji innych genów, takich jak HOXA7 lub HOXA10 jest onkogenne. Co ciekawe białka fuzyjne MLL wydają się być onkogenne wyłącznie dla komórek hematopoetycznych.
Zaburzenia obejmujące gen MLL generalnie są niekorzystnym czynnikiem predykcyjnym odpowiedzi na chemioterapię, choć istnieją doniesienia, że niektóre specyficzne rearanżacje należą do korzystnych lub pośrednich czynników rokowniczych odpowiedzi na leczenie i tym samym na rokowanie.
Translokacja t(1;22) i mutacje GATA-1
Ostra białaczka megakarioblastyczna jest częstsza u dzieci niż u dorosłych. U chorych z zespołem Downa jest związana mutacja GATA-1, a u niemowląt pojawia się translokacja t(1;22)(q13;p13), która prowadzi do powstania białka fuzyjnego OTT-MAL. MAL jest kofaktorem SRF (serum response factor), który posiada silną zdolność aktywacji transkrypcji. Połączenie z genem OTT prowadzi do rozregulowania jego funkcji. OTT jest związany z czynnikiem transkrypcyjnym SHARP, który oddziałuje na szlak Notch/RBP-Jκ. Ekspresja OTT-MAL dereguluje ten szlak, co powoduje deregulację megakariopoezy.
Delecje i zaburzenia liczbowe
Podczas podziałów komórki w wyniku błędów segregacji może dochodzić do utraty lub zyskania dodatkowych chromosomów w komórkach potomnych. Utrata części lub całego chromosomu należy do najczęstszych aberracji spotykanych w ostrej białaczce szpikowej. Delecje są często osiągane w wyniku niezrównoważonych translokacji i utraty części materiału genetycznego. Zmiany zwykle prowadzą do utraty lub unieczynnienia jednego allelu genów supresorowych, ważnych do ochrony komórki przed procesem nowotworzenia. Prawdopodobnie drugi zostaje wyłączony w wyniku kolejnych mutacji lub zmian epigenetycznych. Większość delecji wiąże się z dobrym rokowaniem, choć te złożone wiążą się z agresywnym przebiegiem choroby.
Do najczęstszych delecji w AML należy del 5q, del 7q i del 20q.
Delecja 5q jest dość zróżnicowanym zespołem o wspólnym fenotypie klinicznym. Utrata tej części chromosomu występuje również w zespołach mielodysplastycznych (MDS), w którym dochodzi do ciężkiej niedokrwistości i szybkiej progresji do ostrej białaczki szpikowej. Na długim ramieniu chromosomu 5 występuje kilka genów ważnych dla hematopoezy, w tym genów czynników krwiotwórczych i ich receptorów. Obecnie utrata 9 genów supresorowych zlokalizowanych na tym chromosomie jest podejrzewana o role w powstawaniu ostrej białaczki szpikowej. Są to SSBP2, RIL, APC, SMAD5, EGRI, CTNNA, RPS14, SPARC i NPM.
Częściowa lub całkowita delecja 7q wywołuje różne choroby rozrostowe, szczególnie zespoły mielodysplastyczne i ostrą białaczkę szpikową, a delecja jest obserwowana w różnych guzach litych. Prawdopodobnie krytycznym miejscem jest utrata 7q22 oraz 7q32-q35, jednak nie udało się zidentyfikować żadnego genu odpowiedzialnego za chorobę.
Najczęstszą trisomią spotykaną w AML jest trisomia 8, która pojawia się w 10–15% przypadków ostrej białaczki szpikowej. Trisomia 8 występuje również w MDS oraz kilku guzach litych. Dodatkowa kopia chromosomu 8 prawdopodobnie nie tylko wpływa na ekspresję genów zlokalizowanych w tym chromosomie, ale również globalnie na ekspresję wielu innych genów w innych lokalizacjach. Dochodzi między innymi do zmniejszonej ekspresji genów odpowiedzialnych za apoptozę. Mutacja jest związana ze złym rokowaniem.
Mutacje chromosomalne związane z koaktywatorami transkrypcyjnymi
Opisano kilka mutacji związanych z białkami o funkcji koaktywatora transkrypcji. Należą do nich fuzje MLL/CBP i MOZ/CBP, które dotyczą koaktywatora transkrypcyjnego CBP (CREB binding protein), oraz MLL/p300 i MOZ/TIF2, które obejmują koaktywator p300 i TIF2. TIF2 pełni funkcje rekrutującą CBP, a zatem aktywacja p300 i CBP jest wspólnym elementem obu grup białek fuzyjnych. Cele transkrypcyjne tych białek i ich właściwości nie są jasne. Zmutowane białka powodują zaburzenie różnicowania oraz nieprawidłową proliferację i żywotność komórek.
Zmiany epigenetyczne
Ekspresja genów nie zależy wyłącznie od samego kodu genetycznego zapisanego w sekwencji nukleotydowej w DNA. Zmiany epigenetyczne to dziedziczne modyfikacje regulacji ekspresji genów, która nie zależy od zmian w sekwencji DNA. Obejmują one zmiany metylacji DNA i modyfikację histonów, wpływając na zmiany struktury chromatyny i regulację ekspresji. Zmiany regulacji tych procesów prowadzą do deregulacji struktury chromatyny i zaburzenia ekspresji genów.
Zmiany epigenetyczne współdziałają ze zmianami cytogenetycznymi i mają istotny wpływ na patogenezę ostrej białaczki szpikowej.
Wskazuje się na nieprawidłowy profil metylacji DNA u chorych z AML. Występuje obniżony poziom 5-metylocytozyny (5-mc) z jednoczesną hipermetylacją regionów promotorowych, w szczególności wysp CpG. Skutkuje to wyciszeniem antyonkogenów.
Mutacje IDH1 i IDH2
Mutacje IDH1 i IDH2 są obecne w 5–30% przypadków ostrej białaczki szpikowej. Geny IDH1 i IDH2 kodują dehydrogenazy izocytrynianu 1 i 2 – enzymy zaangażowane w reakcje cyklu Krebsa. IDH1 katalizuje dekarboksylacje izocytrynianu do α-ketoglutaranu (α-KG) i produkcji NADP+. Z kolei IDH2 katalizuje podobną reakcję w mitochondriach.
Zmutowany gen IDH1/2 powoduje katalizuje przemianę α-ketoglutaranu (α-KG) do 2-hydroksyglutaranu (2-HG), który w warunkach prawidłowych występuje w komórkach w bardzo małej liczbie, a w wyniku mutacji jest nadmiernie wytwarzany. Zmieniony poziom α-KG hamuje aktywność enzymów wykorzystujących go jako substrat, które jednocześnie są hamowane kompetytywne przez 2-HG. Hamuje to demetylazy histonów JHDM (Jumonij family of histone demethylases), co ostatecznie prowadzi do hipermetylacji reszt histonowych. 2-HG wpływa na hydroksylazę prolinową, która reguluje czynnik transkrypcyjny HIF-1α, który bierze udział w patogenezie wielu nowotworów.
Mutacje TET2
TET2 należy do rodziny białek TET, które katalizuje reakcję oksydacji 5-metylocytozyny (5-mc) do 5-hydroksymetylocytozyny (5-hmc) w łańcuchu DNA. Proces prawdopodobnie bierze udział w regulacji ekspresji genów. 5-hmc blokuje metylację DNA poprzez blokowanie wiązania białek MDB (methyl-DNA binding proteins) do zmetylowanego DNA i zapobiega wyciszaniu transkrypcji. Jednocześnie konwersja 5-mc do 5-hmc prowadzi do biernej demetylacji DNA.
Mutacje TET2 prowadzą do obniżenia aktywności tego enzymu i spadku stężenia 5-hmc. W badaniach na myszach wykazano, że wyłączenie genu TET2 powoduje zahamowanie różnicowania komórek prekursorowych. Niektóre badania sugerują, że zaburzenie procesu metylacji może być związane z mutacjami IDH1/2, ponieważ TET2 jako enzym zależny od α-KG jest hamowany przez 2-HG, którego podwyższony poziom jest obserwowany przy mutacjach IDH1/2. Prawdopodobnie obie mutacje współdziałają ze sobą w procesie nowotworzenia.
Mutacje DNMT3A
DNMT3A jest metylotransferazą DNA odpowiedzialną za dodawanie grupy metylowej do reszt cytozyny dinukleotydów wysp CpG łańcucha DNA. Zwiększona metylacja wysp CpG wiąże się ze spadkiem ekspresji genów. Mimo że generalnie genom komórek nowotworowych jest hipometylowany, to hipermetylacja w obrębie promotorów wielu genów supresorowych jest powszechna w wielu typach nowotworów. Mutacje DNMT3A najczęściej powodują skrócenie białka oraz spadek zdolności do wiązania DNA, a w konsekwencji spadek zdolności katalitycznych metylotransferazy.
Funkcja i następstwa mutacji DNMT3A nie zostały w pełni wyjaśnione. Niektóre badania sugerują brak istotnych różnic w ekspresji różnych genów, mimo różnic w nasileniu metylacji różnych regionów DNA. Inne wskazują, że mutacja prowadzi do zwiększonej ekspresji genów HOX. Zaobserwowano niższy poziom metylacji genów HOX.
Mutacja DNMT3A występuje u 18–22% chorych na ostrą białaczkę szpikową i należy do niekorzystnych czynników ryzyka.
Mutacje ASXL1
Mutacje genu ASML1 występują w 6–20% przypadków ostrej białaczki szpikowej i są związane z niekorzystnym rokowaniem. Częstość mutacji wyraźnie wzrasta wraz z wiekiem chorych na białaczkę i u chorych powyżej 60. roku życia występują one 4–5-krotnie częściej niż u chorych w wieku 16–60 lat. Biologiczna rola ASXL1 pozostaje mało poznana. Prawdopodobnie wraz z białkiem BAP1 tworzy kompleks usuwający ubikwitynę z histonu H2A. Prawdopodobnie ASXL1 oddziałuje z innymi białkami odpowiedzialnymi za metylację H3K27, hamując transkrypcję.
Mutacje EZH2
EZH2 wraz z białkami SUZ12 i EED współtworzą kompleks PRC2 (grupa białek Polycomb – PcG), który poprzez potrójną metylację histonu H3 prowadzi do wyciszania genów. Nadekspresja EZH2 występuje w wielu zaawansowanych nowotworach i została również zidentyfikowana w ostrej białaczce szpikowej.
Mutacje punktowe
Mutacje punktowe obejmujące pojedyncze geny odgrywają istotną rolę w patogenezie białaczek. Część z tych mutacji pozostaje wciąż nieopisana.
Białka RAS
Białka RAS należą do protoonkogenów, a ich aktywacja do onkogenu jest jedną z najczęstszych zmian genetycznych obserwowanych w nowotworach u ludzi. W ostrej białaczce szpikowej może występować od 10 do 20% wszystkich przypadków tej choroby. Mutacja wiąże się z gorszym rokowaniem. Nadrodzina genów RAS koduje białka błonowe wiążące GTP, które regulują przekazanie sygnału po przyłączeniu ligandu. Regulują wiele procesów komórkowych, w tym różnicowanie, apoptozę, organizacje cytoszkieletu i transport jonowy. Mutacje genów RAS powodują zwiększoną oporność na apoptozę i działanie proproliferacyjne.
Choć mutacje RAS występują dość często w ostrej białaczce szpikowej, to ich rola w patogenezie choroby pozostaje nieustalona. Onkogen RAS prowadzi do stałej stymulacji, która jest niezależna od zewnętrznych bodźców. Część badań wskazuje, że mutacja powstaje na wczesnym etapie karcynogenezy, a inne, że następuje ona w etapie progresji. Mutacja RAS jest potencjalnym miejscem działania leków celowanych.
Mutacje aktywujące kinazę tyrozynową
Kinaza tyrozynowa jest kinazą białkową, która reguluje wiele procesów komórkowych. Aktywująca mutacja kinazy tyrozynowej stanowi kluczowy element patogenezy przewlekłej białaczki szpikowej, gdzie w wyniku translokacji chromosomalnej powstaje białko fuzyjne BCL/Abl o aktywności kinazy tyrozynowej.
Mutacje punktowe aktywujące kinazy tyrozynowe są częste w ostrej białaczce szpikowej. Mutacje powodują trwałą aktywność bez obecności ligandu. Do najczęstszych należy FLT3 i c-KIT, przy czym mutacja FLT3 występuje aż w 30% przypadków ostrej białaczki szpikowej.
Fizjologicznie FLT3 po związaniu ligandu ulega dimeryzacji i zmianom konformacyjnym, następnie aktywuje kinazę tyrozynową i przekazuje sygnał na kolejne mediatory, w których biorą udział między innymi kinaza PI3, AKT, kinaza MAP i STAT5. Ekspresja FLT3 prawidłowo występuje głównie w początkowym stadium hematopoezy w komórkach CD34+. Eksperymentalnie brak FLT3 powoduje znaczne upośledzenie hematopoezy. W ostrej białaczce szpikowej wewnętrzna tandemowa duplikacja (internal tandem duplication, ITD) lub mutacja aktywująca receptora FLT3 powoduje, że nie jest on związany wyłącznie z komórkami CD34+, w niektórych komórkach może występować nadekspresja lub mutacja aktywująca białko FLT3. Wykazano, że mutacje w genie FTL3 wpływają negatywnie na przeżycie całkowite chorych.
Nukleofosmina
Nukleofosmina (NPM) jest fosfoproteiną występującą głównie w jąderku biorącą udział w licznych procesach komórkowych. Bierze udział w transkrypcyjnej modyfikacji rRNA, składaniu rybosomów, stymulując tym samym wzrost i proliferację komórki. NPM wiąże kwasy nukleinowe, przetwarza pre-RNA i pełni rolę białka opiekuńczego. NPM oddziałuje z antyonkogenami, w tym z p53 i ARF. Białko może modulować aktywność p53, gdy czynniki uszkadzające powodują przemieszczenie NPM do cytoplazmy i aktywację p53. Również chroni ARF przed degradacją. Białko wpływa na stabilność genomu poprzez wpływ na naprawę DNA oraz reguluje duplikację centrosomów podczas podziału.
Mutacja NPM1 wydaje się być swoista dla ostrej białaczki szpikowej i tylko sporadycznie opisuje się ją w chłoniakach i zespołach mielodysplastycznych. Jest obserwowana od 25 do 30% przypadków AML u dorosłych. Mutacje genu NPM skutkują nieprawidłową cytoplazmatyczną lokalizacją białka. W efekcie dochodzi do zakłócania różnych sygnałów komórkowych poprzez oddziaływanie zmutowanego białka NPM z innymi białkami i utratę funkcji prawidłowych form NPM w wyniku związania niezmutowanej formy ze zmutowaną i wspólny transport do cytoplazmy. Niektóre badania wskazują, że zmutowana NPM wpływa na zwiększenia poziomu protoonkogenu c-MYC. Zmutowany NPM upośledza zdolność ARF do stabilizowania p53. Cytoplazmatyczny NPM blokuje aktywne kaspazy 6 i 8, co nasila przeżycie komórek białaczkowych.
Istnieją dowody sugerujące, że mutacja NPM może być jedną z pierwotnych mutacji prowadzących do kolejnych wtórnych mutacji, co wskazuje na jej kluczową rolę w patogenezie ostrej białaczki szpikowej.
Białaczkowe komórki macierzyste
W prawidłowym szpiku kostnym istnieje niewielka liczba komórek macierzystych, mających zdolność do samoodnawiania się i do nieograniczonej liczby podziałów, które w wyniku różnicowania dają multipotencjalne hematopoetyczne komórki progenitorowe już pozbawione funkcji samoodnowy, ale mogące ulegać dojrzewaniu do zróżnicowanych komórek krwi obwodowej. Przypuszcza się, że istnieje klon komórek macierzystych posiadający zdolność do nieograniczonego namnażania, który daje białaczkowe komórki progenitorowe bez zdolności do samoodnawiania i bez zdolności do dojrzewania w prawidłowe elementy morfotyczne krwi. Badania na myszach, u których przeszczepiono komórki ludzkiej białaczki wskazują na istnienie populacji komórek zdolnych do samoodnowy, o podobnym immunofenotypie do prawidłowych komórek macierzystych szpiku. Wykazano, że stosunkowo bardzo mała (0,1–0,01% komórek) frakcja komórek CD34+ CD38− wystarcza do odnowy białaczki u myszy. Wskazuje to, że ostra białaczka szpikowa, podobnie jak prawidłowa hematopoeza jest zorganizowana hierarchicznie i niewielka liczba komórek macierzystych wystarcza do wzrostu nowotworu. Dodatkowo wykazano istnienie populacji komórek macierzystych ze zmianami cytogenetycznymi typowymi dla ostrej białaczki szpikowej w prymitywnych komórkach progenitorowych CD34+ CD38−.
Leczenie
Intensywna chemioterapia jest dzielona na trzy fazy: indukcję, konsolidację i podtrzymanie remisji.
Leczenie indukcyjne
Celem leczenia indukcyjnego jest usunięcie widocznych oznak białaczki w szpiku i przywrócenie normalnej hematopoezy. Leczenie indukcyjne rozpoczyna się po zebraniu odpowiedniego materiału do dalszych badań i u większości chorych leczenie można bezpiecznie przełożyć do czasu uzyskania wyników badań genetycznych i molekularnych. Jednak opóźnienie leczenia powyżej 5 dni może przekładać się negatywnie na wyniki terapii.
Wytyczne NCCN zalecają jeden z trzech programów leczniczych:
- DA 3+7 – przez pierwsze 3 dni stosuje się antracyklinę: daunorubicynę w dawce co najmniej 90 mg/m² lub idarubicynę w dawce 12 mg/m², a następne przez kolejne 7 dni podaje się cytarabinę w standardowej dawce 100–200 mg/m²,
- DAC – przez pierwsze 3 dni podaje się daunorubicynę w dawce 60 mg/m², cytarabinę w dawce 200 mg/m² 1–7 dnia cyklu oraz kladrybinę w dawce w 5 mg/m² w 1–5 dniu cyklu,
- HiDAC (wysoko dawkowana cytarabina) – stosuje się cytarabinę w dawce 2 g/m² co 12h przez 6 dni lub cytarabinę w dawce 3 g/m² co 12h przez 4 dni oraz idarubicynę w dawce 12 mg/m² albo daunorubicynę w dawce 60 mg/m².
Wykazano, że wyższa dawka daunorubicyny (90 mg/m²) wywołuje wyższy odsetek odpowiedzi całkowitych i wydłuża przeżycie całkowite w porównaniu do standardowej dawki (90 mg/m²) daunorubicyny, która była stosowana w dotychczasowych schematach. W tym badaniu największe korzyści z wysokiej dawki cytarabiny uzyskali chorzy w grupie wysokiego ryzyka, w mniejszym stopniu chorzy w grupie pośredniego ryzyka, z kolei korzyści u chorych o niskim ryzyku były ograniczone.
W dużym polskim badaniu wykazano, że dodanie kladrybiny do daunorubicyny (60 mg/m²) z cytarabiną (200 mg/m²) powoduje znacząco większą odpowiedź całkowitą (67,5% z kladrybiną, 56% bez kladrybiny) oraz zwiększone trzyletnie przeżycie całkowite (45% z kladrybiną, 33% bez kladrybiny). Schemat DAC wiąże się z lepszym przeżyciem całkowitym również u chorych w grupie wysokiego ryzyka.
W badaniu SWOG oceniono leczenie indukcyjne za pomocą wysokich dawek cytarabiny (HiDAC). W tym badaniu leczeni wysoką dawką cytarabiny osiągnęli podobne przeżycie wolne od choroby oraz przeżycie całkowite w porównaniu do standardowej dawki cytarabiny, jednocześnie leczenie wysokimi dawkami cytarabiny było związane z wyższą śmiertelnością związaną z leczeniem. W związku z porównywalnymi efektami stosowanie wysokiej dawki cytarabiny w leczeniu indukcyjnym jest kontrowersyjne. Jednak decyzja o zastosowaniu wysokiej dawki może być konieczna przed zastosowaniem autogenicznego przeszczepienia szpiku (allo-HCT), ponieważ dwa badania wskazują na szybsze usunięcie blastów po jednym cyklu chemioterapii i wyższe przeżycie wolne od choroby u leczonych poniżej pięćdziesiątego roku życia.
Odsetek remisji jest uzależniony od grupy ryzyka cytogenetycznego. Remisję osiąga nawet 87% chorych ze zmianami cytogenetycznymi korzystnego ryzyka, 79% ze zmianami pośredniego ryzyka i 62% w grupie niekorzystnego ryzyka.
Badano czynniki wzrostu G-CSF i GM-CSF pod kątem zwiększenia wrażliwości komórek białaczkowych na leczenie. Część badań wskazuje na korzyści wynikające ze stosowania G-CSF, a część ich nie potwierdza.
Chorzy z nadmierną leukocytozą i objawami klinicznymi leukostazy mogą wymagać leukaferezy, która jest koordynowana z rozpoczęciem chemioterapii. Są to chorzy o szczególnym ryzyku wystąpienia zespołu ostrego rozpadu guza i wymagają odpowiedniego monitorowania.
Ocena skuteczności
Ocena skuteczności terapii indukcyjnej następuje po 7–10 dniach, wykonuje się wówczas biopsję aspiracyjną szpiku. Zwykle stwierdza się niską komórkowość szpiku (<20%) i mały odsetek blastów (5–10%), potwierdza to skuteczność leczenia indukcyjnego. Przed dalszym leczeniem należy poczekać na regenerację hematopoezy, co zwykle trwa około 3–4 tygodni i następnie powtarza się biopsję szpiku celem potwierdzenia remisji.
Stwierdzenie w pierwszej biopsji dużego odsetka wskazuje na oporność i zmusza do modyfikacji leczenia.
Leczenie w nieskuteczności chemioterapii indukcyjnej
W przypadku nieskuteczności leczenia indukcyjnego dalsze postępowanie zależy od stopnia uzyskanej odpowiedzi. W przypadku występowania częściowej remisji (PR) i występowania rezydualnych blastów można rozważyć podanie dodatkowego cyklu leczenia w standardowej dawce antracykliny i cytarabiny. Dla większej liczby blastów można zastosować cytarabinę w wysokich dawkach (HiDAC) lub cytarabinę w standardowych dawkach z antracykliną. W przypadku niepowodzenia leczenia wysokimi dawkami cytarabiny opcjami są allo-HCT, udział w badaniach klinicznych lub schematy ratujące.
Konsolidacja remisji
Konsolidacja jest uzasadniona u chorych, którzy osiągnęli remisję hematologiczną i kliniczną.
Celem leczenia konsolidacyjnego jest likwidacja pozostałej populacji komórek nowotworowych, czyli choroby resztkowej. Jest to populacja, którą można rozpoznać tylko w bardzo czułych badaniach, takich jak cytometria przepływowa czy w metodach molekularnych. Przetrwałe komórki mogą być bardziej oporne na leczenia, dodatkowo mogą występować w strefach o ograniczonej zdolności penetracji leków. Leczenie jest powtarzane w regularnych cyklach, tak by zniszczyć komórki znajdujące się w tych etapach cyklu komórkowego, w których są niewrażliwe na cytostatyki. Leczenie jest uzależnione od określonej grupy ryzyka chorych.
Grupa korzystnego ryzyka
W grupie korzystnego ryzyka, jeśli nie ma wskazań do allo-HCT zaleca się podanie 3–4 cykli HiDAC.
W badaniu CALGB 4-letnie przeżycie bez objawów choroby (DFS) chorych leczonych cytarabiną w wysokiej dawce 4 g/m² wynosiło 44%. W tym innym badaniu wykazano, że przeżycie wolne od nawrotu choroby (RFS) w różnych grupach cytogenetycznych jest odmienne. W grupie chorych z normalnym kariotypem 5-letnie RFS wynosiło 32%, a z nieprawidłowościami cytogenetycznymi 15%.
Badanie HOVON/SAKK sugeruje, że dwa cykle pośredniej dawki cytarabiny mogą być alternatywą dla trzech cykli cytarabiny w wysokiej dawce.
Alternatywnym postępowaniem jest wysoka dawka lub zwiększona liczba cykli cytarabiny, a następnie autogeniczne (auto-HCT) lub allogeniczne przeszczepienie szpiku (allo-HCT) od spokrewnionego lub niespokrewnionego dawcy (szczególnie w mutacji C-KIT). Wybór najkorzystnej metody leczenia uwzględnia ryzyko nawrotu białaczki oraz ryzyko związane z procedurą przeszczepu.
Grupa pośredniego ryzyka
U chorych z grupy pośredniego i wysokiego ryzyka zakwalifikowanych do allo-HCT konsolidacja powinna osiągnąć jak najlepszą całkowitą remisję i nie opóźniać transplantacji.
W grupie pośredniego ryzyka stosuje się 3–4 cykli HiDAC, jednak wyniki są niezadowalające. Wykazano korzyści przeszczepienia szpiku (allo-HCT) u chorych z pośrednim ryzykiem cytogenetycznym. Przeszczepienie szpiku (allo-HCT) jest korzystniejsze dla osób z wysokim ryzykiem nawrotu i niskim lub pośrednim ryzykiem przeszczepu. Z kolei wysokodawkowa chemioterapia i autogeniczne przeszczepienie szpiku jest postępowaniem kontrowersyjnym w grupie pośredniego ryzyka. Część danych sugeruje, że może być ono alternatywą dla przeszczepienia autogenicznego, jednak choć prawdopodobnie wydłuża okres remisji, to zdolność do wydłużenia przeżycia całkowitego jest niepewna. Zalecenia NCCN nie zalecają go poza badaniami klinicznymi.
Grupa wysokiego ryzyka
Leczenie jest oparte na allogenicznym przeszczepie szpiku od dawcy spokrewnionego lub niespokrewnionego. Konsolidacja do czasu uzyskania zgodnego dawcy jest oparta na cytarabinie w wysokiej dawce.
Leczenie pokonsolidacyjne (podtrzymujące)
Leczenie po konsolidacji służy utrwaleniu stanu remisji. W grupie korzystnego ryzyka po konsolidacji zaleca się jedynie monitorowanie choroby resztkowej. W przypadku stwierdzenia nawrotu przeprowadza się ponowną indukcje remisji i następnie wykonuje się allogeniczne przeszczepienie szpiku. U chorych w grupie pośredniego i wysokiego ryzyka w ramach konsolidacji zaleca się przeszczepienie szpiku.
Przeszczepienie szpiku
Allogeniczne przeszczepienie szpiku

Allogeniczne przeszczepienie szpiku (allo-HCT) jest strategią leczenia poremisyjnego związanego z najniższym odsetkiem nawrotu, jednak korzyści wynikające z tej terapii są ograniczane przez śmiertelność związaną z leczeniem. Efekt przeciwbiałaczkowy wynika z wysokodawkowej chemioterapii stosowanej w kondycjonowaniu (leczenie mające na celu zniszczenie szpiku kostnego biorcy) oraz reakcji przeszczep przeciw białaczce (graft versus leukemia – GvL).
Allogeniczne limfocyty T rozpoznają antygeny białaczkowe specyficzne i towarzyszące, a także niezgodności słabych antygenów zgodności tkankowej (MHA) jako obcy antygen, co skutkuje odpowiedzią immunologiczną na antygeny białaczkowe w reakcji przeszczep przeciw białaczce. Jednocześnie dochodzi do rozwoju niekorzystnej reakcji przeszczep przeciw gospodarzowi (graft-versus-host disease, GvHD). Najczęściej korzyściom wynikającym z reakcji przeciw białaczce towarzyszy reakcja przeciw gospodarzowi, wywołując uszkodzenia głównie w skórze, przewodzie pokarmowym, wątrobie i płucach. Niekontrolowana reakcja GvHD może doprowadzić do śmierci biorcy przeszczepu. Z drugiej strony chorzy z GvHD, szczególnie z postacią przewlekłą choroby, rzadziej rozwijają nawroty ostrej białaczki szpikowej. W przyszłości nowoczesna immunoterapia może pomóc wykorzystać zjawisko GvL bez niekorzystnej reakcji GvHD.
Istotnym problemem jest śmiertelność towarzysząca temu leczeniu (transplant-related mortality – TRM), która wynosi około 15–20%. Śmiertelność jest związana głównie z chorobą przeszczep przeciwko gospodarzowi oraz powikłaniami wynikającymi z immunosupresji.
Bardzo ważna jest odpowiednia kwalifikacja kandydatów do przeszczepu, w której korzyści wynikające z leczenia przewyższają ryzyko związane z procedurą. W ocenie ryzyka śmiertelności bierze się pod uwagę obecne zmiany genetyczne, typ dawcy (spokrewniony, niespokrewniony), zastosowanie kondycjonowania o zmniejszonej intensywności (RIC), wiek, współistniejące choroby, stadium choroby, czas od rozpoznania do transplantacji, rodzaj dawcy i dopasowanie pomiędzy dawcą i biorcą. W ocenie pomocna może być skala HCTCI.
Allogeniczne przeszczepienie szpiku jest wskazane u:
- chorych w pierwszej całkowitej remisji (CR1) z grupy dużego i pośredniego ryzyka w dobrym stanie klinicznym,
- chorych w drugiej (CR2) i kolejnych całkowitych remisjach we wszystkich grupach ryzyka,
- chorych w częściowej remisji (PR) lub we wczesnym okresie wznowy.
Metaanaliza badań klinicznych porównująca allogeniczne przeszczepienie szpiku z innymi metodami terapii konsolidacyjnej wskazuje, że daje ono znaczne korzyści chorym o pośrednim lub wysokim ryzyku.
Przeszczep uzyskany od zgodnego dawcy niespokrewnionego zapewnia porównywalne, choć gorsze, wyniki leczenia z dawcą spokrewnionym. W jednym badaniu wykazano, że 3-letnie przeżycie wolne od choroby po przeszczepie od dawcy spokrewnionego wynosiło 34%, a od niespokrewnionego 42%. Podobne wyniki osiąga się również u chorych o pośrednim ryzyku. Najdłuższe przeżycie całkowite osiągają chorzy o pełnej zgodności pomiędzy dawcą i biorcą przeszczepu od dawcy niespokrewnionego w układzie HLA.
Allogeniczne przeszczepienie szpiku jest jedną z możliwych opcji terapeutycznych u chorych w grupie korzystnego ryzyka. Kilka badań sugeruje korzyści wynikające z niego w grupie o korzystnym ryzyku, szczególnie u chorych poniżej 55. roku życia.
Procedura allo-HCT u chorych bez całkowitej remisji pozostaje kontrowersyjnym zagadnieniem. Problem aktywnej choroby obejmuje zróżnicowaną grupę chorych z pierwotną opornością, wczesnym nawrotem, oporną wznową, czy kolejnymi wznowami. Badanie CIBMTR wykazało 19% odsetek 3-letniego przeżycia całkowitego u chorych po przeszczepie bez całkowitej remisji. Prawdopodobnie leczenie sekwencyjne z chemioterapią poprzedzającą przeszczepienie szpiku poprawia wyniki; na taki efekt wskazuje wieolośrodkowe badanie, w którym trzyletnie przeżycie całkowite po przeszczepieniu osiągnęło 30% leczonych.
Autogeniczne przeszczepienie szpiku
Autogeniczne przeszczepienie szpiku może być uważane za alternatywę dla allogenicznego przeszczepienia szpiku w postępowaniu poremisyjnym dla chorych o korzystnym lub pośrednim ryzyku, jednak nie jest zalecane u chorych z grupy wysokiego ryzyka. Zaletą metody jest niska śmiertelność związana z leczeniem, jednak towarzyszy temu stosunkowo wysoki odsetek nawrotów. Wyniki po autogenicznym przeszczepieniu szpiku są podobne do leczenia wysokimi dawkami cytarabiny, jednak nie ma dowodu na poprawę przeżycia.
Leczenie nawrotów choroby

Postępowanie w nawrocie choroby jest odmienne w zależności od okresu, który upłynął od remisji. W przypadku późnego nawrotu (według wytycznych NCCN jest to nawrót po 12 mięsiącach od uzyskania remisji całkowitej) stosuje się ten sam schemat co w indukcji remisji. Wczesne remisje traktuje się jak przypadki oporne i stosuje się schematy ratujące. Po uzyskaniu remisji należy dążyć do przeszczepu, ponieważ kolejne remisje są mniej trwałe.
Schematy ratujące zalecane przez NCCN:
- kladrybina z cytarabiną i G-CSF z lub bez mitoksantonem lub idarubicyną,
- wysoka dawka cytarabiny i antracykliny,
- etopozyd i cytarabina z lub bez mitoksantonem,
- klofarabina z lub bez cytarabiny i G-CSF z lub bez idarubicyny.
Leczenie o zmniejszonej toksyczności
Chorzy w starszym wieku lub ze współistniejącymi poważnymi schorzeniami często nie kwalifikują się do leczenia, dlatego wymagają oni osobnej strategii leczniczej.
- Indukcja remisji u chorych powyżej 60. roku życia
U chorych powyżej 60. roku życia leczenie indukcyjne jest uzależnione od stanu ogólnego chorego, które jest oceniane w skali ECOG, a także od obecności niekorzystnych mutacji, występowania zespołu mielodysplastycznego, białaczki wtórnej do leczenia oraz współwystępujących istotnych chorób. Sam wiek nie jest wyłącznym kryterium wyboru opcji terapeutycznych.
Chorzy w stopniu PS0-PS2 bez istotnych chorób współistniejących i bez niekorzystnych mutacji genetycznych mogą być leczeni:
- standardową dawką cytarabiny z idarubicyną lub daunorubicyną albo mitroksantronem,
- chemioterapią o małej intensywności: podskórne podanie cytarabiny lub azacytydyną albo decytabiną.
Chorzy w stopniu PS0-PS2 bez istotnych chorób współistniejących z mutacjami wysokiego ryzyka, białaczką wtórną do leczenia lub zespołu mielodysplastycznego mogą być leczeni:
- standardową dawką cytarabiny z idarubicyną lub daunorubicyną albo mitroksantronem, szczególnie gdy chory jest kandydatem do przeszczepienia szpiku,
- chemioterapią o niskiej intensywności z podskórnym podaniem cytarabiny lub z azacytydyną albo decytabiną, szczególnie gdy chory nie toleruje standardowej chemioterapii indukcyjnej.
Chorzy w złym stanie ogólnym (≥PS3) lub chorzy z istotnymi chorobami towarzyszącymi mogą być leczeni:
- chemioterapią o niskiej intensywności: azacytydyna lub decytabina albo podskórnie cytarabina,
- najlepsza terapia wspomagająca, w tym hydroksymocznik i transfuzje preparatów krwiopochodnych.
Leczenie indukcyjne u chorych powyżej 60. roku życia w dobrym stanie ogólnym, czyli poniżej PS2, bez istotnych chorób współistniejących składa się z terapii antracykliną (daunorubicyna w 45–60 mg² lub innej antracykliny w równoważnej dawce) przez 3 dni, a następnie przez 7 dni cytarabina w dawce 100–200 mg². Kilka badań wykazało, że antracykliny nie wykazują pomiędzy sobą znaczących różnic w leczeniu indukcyjnym chorych powyżej 60. roku życia. Jednak dwa francuskie badania wskazują na pewną przewagę idarubicyny u chorych powyżej 50. roku życia, dlatego w zaleceniach NCCN idarubicyna jest preferowana wśród pozostałych antracyklin w leczeniu indukcyjnym chorych powyżej 60. roku życia. Badanie HOVON/SAKK/AMLSG wskazuje, że daunorubicyna w wysokiej dawce (3×90 mg²) powoduje większy odsetek całkowitych remisji i dłuższe przeżycie całkowite u chorych powyżej 60. roku życia.
Występowanie niekorzystnych mutacji cytogenetycznych jest silnym i niezależnym czynnikiem predykcyjnym osiągnięcia całkowitej remisji i osiąganego przeżycia całkowitego.
Dla chorych nienadających się do standardowej terapii indukcyjnej stosuje się chemioterapię o niskiej intensywności, która obejmuje niskie dawki cytarabiny, azacytydynę lub decytabinę.
Azacytydyna zwiększa przeżycie całkowite w porównaniu z najlepszą terapią wspomagającą, z małą dawką cytarabiny lub intensywną chemioterapią. Mediana przeżycia całkowitego wynosiła 24,4 miesiąca dla leczonych azacytydyną i 16 miesięcy dla leczonych standardowym leczeniem.
Decytabina w jednym badaniu wywoływała 24% odsetek odpowiedzi całkowitych. W innym badaniu lek wykazywał lepszy odsetek odpowiedzi całkowitej w porównaniu do małych dawek cytarabiny lub intensywnej chemioterapii, jednak nie zwiększał znacząco przeżycia całkowitego.
Chorzy w złym stanie ogólnym i istotnymi chorobami współistniejącymi są bardziej narażeni na efekty toksyczne leczenia oraz osiągają mniejsze korzyści ze standardowego leczenia indukcyjnego. Dla tych pacjentów zalecana jest chemioterapia o niskiej intensywności lub najlepsza opieka wspomagająca.
- Konsolidacja remisji u chorych powyżej 60. roku życia
U chorych powyżej 60. roku życia zalecenia NCCN przewidują konsolidację remisji za pomocą:
- allogenicznego przeszczepienia szpiku (allo-HCT) o obniżonej toksyczności,
- antracykliny i cytarabiny w standardowej dawce 100–200 mg/m² („schemat 3+7”),
- cytarabiny 1–1,5 g/m² u chorych w dobrym stanie ogólnym, prawidłową funkcją nerek i korzystnym lub prawidłowym kariotypie,
- programów leczniczych o małej intensywności z azacytydyną lub decytabiną.
Badanie CALGB porównało 2 schematy leczenia dla osób powyżej 60. roku życia i nie wykazano różnicy pomiędzy intensywnym leczeniem cytarabiną (500 mg/m²) z mitoksantronem a cytarabiną w standardowej dawce (100 mg/m²). Badanie ALFA 98 wykazało wyższość 6 ambulatoryjnych cykli cytarabiny (60 mg/m²) i antracyklin nad 1 intensywnym cyklem antacyklin i cytarabiny (200 mg/m²) w schemacie „4+7”. Szwedzkie badanie na rejestrach sugeruje, że u chorych w dobrym stanie ogólnym i brakiem istotnych chorób współistniejących schemat „3 + 7" zastosowany w indukcji może być użyty w mniejszych dawkach.
Allogeniczne przeszczepienie szpiku ma ograniczoną rolę w leczeniu osób powyżej 65. roku życia lub ze współistniejącymi chorobami. Jednak obiecujące wyniki dają badania nad przeszczepianiem szpiku o zmniejszonej intensywności kondycjonowania (reduced-intensity conditioning, RIC). Kondycjonowanie jest to leczenie za pomocą megachemioterapii lub radioterapii, która ma na celu zniszczenie szpiku kostnego, co poprzedza przeszczepienie.
Niektóre serie przypadków sugerują wydłużenie przeżycia całkowitego po przeszczepieniu szpiku ze zmniejszoną intensywnością kondycjonowania w porównaniu ze standardowym kondycjonowaniem. Z kolei badanie retrospektywne porównujące chorych 50–70-letnich, którzy otrzymali przeszczep szpiku (metodą o standardowym kondycjonowaniu oraz RIC) z chorymi, którzy nie otrzymali przeszczepu wskazuje na wyższe przeżycie wolne od nawrotu choroby (RFS), wyższe 3-letnie przeżycia całkowite u chorych po przeszczepie i mniejszy odsetek nawrotów choroby. W badaniu większość chorych z przeszczepem szpiku przeszła procedurę o zmniejszonej intensywności kondycjonowania. W innym badaniu porównano RIC ze standardową chemioterapią indukcyjną i poindukcyjną. Chory po allogenicznym przeszczepie wykazywali dłuższe przeżycie wolne od choroby oraz niższy odsetek nawrotów, jednak przeżycie całkowite nie różniło się istotnie między grupami. Według zaleceń NCCN allogeniczne przeszczepienie szpiku o zmniejszonej intensywności kondycjnowanie (RIC) jest opcją terapeutyczną w leczeniu poremisyjnym dla chorych powyżej 60. roku życia, szczególnie przy dobrym stanie ogólnym.
Leczenie ostrej białaczki promielocytowej z t(15;17)

Ostra białaczka promielocytowa (APL) jest pierwszym podtypem, który można opanować poprzez pobudzenie zablokowanego dojrzewania i indukcję apoptozy. W celu zmniejszenia wczesnej śmiertelności z powodu koagulopatii leczenie APL jest włączane niezwłocznie przed ostatecznym rozpoznaniem molekularnym, po wstępnym rozpoznaniu APL na podstawie morfologii, immunofenotypowania lub występowania zespołu rozsianego wykrzepiania wewnątrznaczyniowego. Do ostatecznego rozpoznania jest wymagane badanie molekularne.
- Leczenie indukcyjne APL
W leczeniu indukcyjnym stosuje się kwas całkowicie trans-retinowy (ATRA, od ang. all-trans retinoic acid) oraz antracykliny. Schemat prowadzi do wystąpienia całkowitej remisji u aż 90–95% leczonych, a oporność jest wyjątkowo rzadka.
ATRA nie może być podawana w monoterapii, bo choć samodzielnie wywołuje znaczny odsetek remisji, to szybko dochodzi do rozwoju nawrotu, dlatego ATRA jest łączona z innymi lekami, przede wszystkim antracyklinami. Wybór optymalnej antracykliny i czy antracyklinę należy łączyć z innym lekiem jest przedmiotem badań klinicznych.
Porównywalny odsetek całkowitych remisji uzyskano za pomocą ATRA z daunorubicyną i z cytarabiną oraz ATRA z idurubicyną. Wstępne wyniki randomizowanego badania nie wykazują różnic między ATRA z idarubicyną a ATRA z daunorubicyną i cytarabiną. Część badań sugeruje efekty hiperaddycyjne dodania cytarabiny do ATRA w połączeniu z antracyklinami.
Trójtlenek arsenu (ATO) jest silnym promotorem apoptozy w promieloblastów.
ATO wykazuje podobnie wysokie wyniki w leczeniu APL, odsetek całkowitej odpowiedzi przekraczający 90%, zarówno w monoterapii, jak i w połączeniu z ATRA. Jednak w połączeniu z ATRA zaobserwowano bardziej nasilone obniżenie liczby transkryptów PML/RARα. Randomizowane badanie wykazało równoważność ATRA w połączeniu z trójtlenkiem arsenu ze schematem AIDA (ATRA i idarubicyna) u chorych z niskim lub pośrednim ryzykiem. Niektóre badania wskazują na korzyści w leczeniu chorych o dużym ryzyku.
Dla chorych o niskim lub pośrednim ryzyku (liczba krwinek białych <10 000/μl) wytyczne NCCN zalecają stosowanie ATRA w połączeniu z trójtlenkiem arsenu (ATO), ewentualnie ATRA z daunorubicyną i cytarabiną, trzecią opcją jest ATRA i idarubicyna (schemat AIDA). Dla chorych o wysokim ryzyku stosuje się połączenie ATRA z cytarabiną i daunoruicyną, połączenie ATRA z idarubicyną, połączenie ATRA z idarubicyną i trójtlenkiem arsenu.
- Konsolidacja remisji APL
Po 6–8 tygodniach od przeprowadzenia leczenia indukcyjnego pobiera się szpik kostny celem oceny skuteczności leczenia. Zwykle badanie cytogenetyczne jest prawidłowe, ale uzyskanie remisji molekularnej wymaga leczenia konsolidacyjnego i ocena remisji molekularnej jest przeprowadzana po leczeniu konsolidacyjnym.
W drugim badaniu PETHEMA wykazano, że dodanie ATRA do konsolidacji opartej na antracyklinach w grupie wysokiego ryzyka zmniejsza odsetek nawrotów (z 20,1% do 8,7%), w grupie o pośrednim ryzyku dodanie również ATRA zmniejszała odsetek nawrotów (14% do 2,5%), ale w grupie o niskim ryzyku dodanie leku nie dawało dodatkowych korzyści. W badaniu PETHEMA LPA 2005 porównano różne schematy leczenia z ATRA i cytarabiną, badanie wskazuje, że połączenie ATRA, cytarabiny oraz idarubicyny zmniejsza odsetek nawrotów w grupie wysokiego ryzyka. Również badanie GIMEMA AIDA-2000 potwierdza, że ATRA z cytarabiną i idarubicyną redukuje odsetek nawrotów.
W badaniu APL 2000 wykazano, że chorzy otrzymujący w konsolidacji remisji cytarabinę w połączeniu z daunorubicyną wykazują mniejszy odsetek nawrotów w grupie wysokiego ryzyka w porównaniu do chorych otrzymujących samą daunorubicynę.
Amerykańskie badanie wskazuje na skuteczność zastosowania w konsolidacji schematu składającego się z 2 cykli ATRA w połączeniu z ATO, a następnie 2 cykli ATRA i daunorubicyny, który porównano do leczenia wyłącznie ATRA i daunorubicyną. Schemat z ATO wykazał wyższe przeżycie wolne od choroby (90% z ATO, 70% bez ATO) i trzyletnie przeżycie całkowite (86% z ATO, 81% bez ATO), a korzyści były obserwowane we wszystkich grupach ryzyka. Wyniki nie są znacząco lepsze od mniej skomplikowanych schematów dla grupy niskiego i pośredniego ryzyka, jednak w grupie wysokiego ryzyka schemat poprawia przeżywalność.
W badaniu APL0406 u chorych o niskim lub pośrednim ryzyku badano połączenie ATRA i ATO w leczeniu konsolidacyjnym, które porównano z ATRA z antacyklinami. Wykazano przedłużenie odsetka dwuletniego przeżycia całkowitego, bez wydłużenia przeżycia wolnego od choroby i odsetka nawrotów.
Wytyczne NCCN dla chorych o niskim lub pośrednim ryzyku zalecają stosowanie w konsolidacji ATRA w połączeniu z ATO. Wytyczne NCCN dla chorych o wysokim ryzyku zalecają stosowanie w konsolidacji połączenia cytarabiny z ATRA z idarubicyną, połączenia cytarabiny i daunorubicyny (ATRA tylko w indukcji), połączenia 2 cykli ATO, z kolejnymi 2 cyklami standardowej chemioterapii, a w przypadku nietolerancji antracyklin połączenia ATO z ATRA.
- Leczenie podtrzymujące APL
Po osiągnięciu remisji chorzy są oceniani pod kątem osiągnięcia remisji molekularnej. Jeśli leczony osiągnął remisję molekularną może otrzymać 1–2-letnie leczenie podtrzymujące za pomocą ATRA. Leczenie podtrzymujące może być połączone z 6-merkaptopuryną i metotreksatem.
Profilaktyka ośrodkowego układu nerwowego
Zajęcie OUN w momencie rozpoznania występuje u 5% chorych. Obecnie nie zaleca się dokanałowej profilaktyki bez objawów, ale może być rozważona w przypadku znacznej hiperleukocytozy.
W przypadku zajęcia OUN podaje się dokanałowo 40–50 mg cytarabiny 2 lub 3 razy w tygodniu do czasu oczyszczenia z blastów, a następnie trzy dalsze podania w tej samej dawce. W celu zapobiegnięcia zapaleniu pajęczynówki podaje się 4 mg trzy razy dziennie deksametazonu. U chorych z nawrotami może być przydatna radioterapia lub dooponowe podanie leków.
Leczenie wspomagające


W leczenie ostrej białaczki szpikowej istotne jest leczenie wspomagające. Leczenie wspomagające, choć nie może wyleczyć białaczki, to często poprawia przeżywalność chorych, zapobiega ostrym powikłaniom choroby i poprawia jakość życia chorych.
Leczenie małopłytkowości w przebiegu ostrej białaczki szpikowej za pomocą koncentratu krwinek płytkowych (KPP) stosuje się wyłącznie w przypadku krwotoku. Profilaktycznie podaje się koncentrat krwinek płytkowych przy liczbie płytek poniżej 10 000 tys./mm³.
W przypadku niedokrwistości ze stężeniem hemoglobiny poniżej 8 g/dl podaje się koncentrat krwinek czerwonych. Nie ma dowodów na skuteczność podawania koncentratu granulocytarnego.
Podaje się preparaty ubogoleukocytarne, aby zapobiec alloimmunizacji i transmisji zakażenia cytomegalowirusem, który jest potencjalnie groźny dla chorych z upośledzoną odpornością bez przebytego zakażenia. Napromieniowane preparaty mogą zapobiec TA-GVHD. Nie wypracowano konsensusu w sprawie momentu włączenia napromieniowanych preparatów krwiopochodnych, zwykle stosuje się je od momentu kondycjonowania do 6 miesięcy po allogenicznym przeszczepie szpiku, a także 7 dni przed kolekcją komórek macierzystych i 3 miesiące po allogenicznym przeszczepie.
- Czynniki wzrostu
Liczne badania wykazały, że czynniki wzrostu (GM-CSF i G-CSF) przyspieszają regenerację liczby neutrofili od 2 do 5 dni i mogą zmniejszać zapotrzebowanie na antybiotyki, czas trwania gorączki neutropenicznej bez negatywnego wpływu na regenerację liczby płytek krwi i stymulacji namnażania komórek białaczkowych. Jednak czynniki wzrostu nie przekładają się na poprawę przeżycia całkowitego. Czynniki wzrostu należy rozważyć w przypadku ciężkich zakażeń przy jednoczesnej neutropenii.
- Profilaktyka zakażeń
Choroby infekcyjne są przyczyną znacznej śmiertelności u chorych z neutopenią. Istotna jest profilaktyka zakażeń, w tym poprzez profilaktyczne stosowanie chemioprofilaktyki przeciwbakteryjnej i przeciwgrzybiczej. W prewencji stosuje fluorochinolony (ciprofloksacynę, lewofloksacynę), jednak wybór antybiotyku jest również uwarunkowany profilem występujących patogenów i opornością na antybiotyki. Wykazano, że profilaktyka antybiotykami zmniejsza śmiertelność u chorych z neutopenią.
Stosuje się profilaktykę zakażeń grzybiczych, szczególnie za pomocą leków aktywnych wobec Aspergillus. Do tego celu wykorzystuje się itrakonazol, posakonazol i amfoterycyna B. Profilaktyka przeciwgrzybicza prawdopodobnie zmniejsza śmiertelność chorych z neutopenią.
- Znaczna hiperleukocytoza
Znaczna hiperleukocytoza jest definiowana jako liczba leukocytów we krwi powyżej 100 000/μl. Jest ona związana ze znaczną śmiertelnością z powodu powikłań krwotocznych, zespołu lizy guza i powikłań infekcyjnych. Również z powodu leukostazy znaczna hiperlekocytoza wymaga szybkiego leczenia. Zaleca się podawanie hydroksymocznika w dawce 50–60 mg dziennie, tak aby obniżyć liczbę leukocytów do 20 000/μl. Przydatna w obniżeniu nadmiernej leukocytozy może być leukafereza. Jednocześnie należy wdrożyć profilaktykę zespołu lizy guza poprzez odpowiednie nawodnienie i allopurynol.
Kryteria remisji
| Kryterium | Remisja całkowita (CR) | Remisja częściowa (PR) | Brak remisji (NR) |
| Stan ogólny | pełna sprawność | znaczna poprawa | brak poprawy |
| Zmiany pozaszpikowe | nieobecne | zmniejszenie | bez poprawy |
| Blasty w szpiku | <5% – czułe metody mogą wykrywać choroby resztkowej | 5–20% spadek o 50% <5% gdy obecne są pałeczki Auera |
>20% |
| Blasty we krwi obwodowej | 0 | 0 | obecne |
| Granulocyty | >1000/μl | >1000/μl | bez istotnej poprawy |
| Trombocyty | >100 000/μl | >100 000/μl | bez istotnej poprawy |
| Erytrocyty | niezależność od przetoczeń KKCz |
Czynniki prognostyczne i stratyfikacja ryzyka
Rokowanie chorego zależy od stanu jego zdrowia i czynników ryzyka związanych z chorobą
Czynniki ryzyka związane z chorym
Starszy wiek jest najważniejszym czynnikiem prognostycznym związanym z chorym. Chorzy w podeszłym wieku osiągają gorsze wyniki leczenia niezależnie od innych czynników ryzyka. Sam wiek nie jest jednak powodem do dyskwalifikacji z leczenia, a także nie jest najważniejszym czynnikiem ryzyka śmiertelności związanej z leczeniem oraz oporności na leczenie. Kluczowe jest wydzielenie grupy chorych, którzy osiągną korzyści z intensywnej chemioterapii. Bardzo istotne są choroby współistniejące i stan ogólny chorego.
Czynniki ryzyka związane z chorobą
Czynniki ryzyka związane z chorobą obejmują liczbę krwinek białych, wcześniejszy zespół mielodysplastyczny, wcześniejsze leczenie przeciwnowotworowe, ale przede wszystkim zależą od zmian cytogenetycznych i molekularnych. Kariotyp komórek nowotworowych jest najsilniejszym czynnikiem prognostycznym odpowiedzi na leczenie. Wytyczne European LeukemiaNet wyróżniają 4 grupy ryzyka cytogenetycznego w zależności od rokowania: korzystne, pośrednie I, pośrednie II i niekorzystne.
| Grupa ryzyka | Zaburzenia genetyczne |
| Korzystne | t(8;21)(q22;q22) – RUNX1-RUNX1T1 inv(16)(p13.1q22) lub t(16;16)(p13.1;q22) – CBFB-MYH11 mutacja NPM1 bez FLT3-ITD (prawidłowy kariotyp) mutacja CEBPA (prawidłowy kariotyp) |
| Pośrednie I | Mutacja NPM1 and FLT3-ITD (prawidłowy kariotyp) wtNPM1 i FLT3-ITD (prawidłowy kariotyp) wtNPM1 bez FLT3-ITD (prawidłowy kariotyp) |
| Pośrednie II | t(9;11)(p22;q23) – MLLT3-MLL nieprawidłowości cytogenetyczne niesklasyfikowane jako korzystne lub niekorzystne |
| Niekorzystne | inv(3)(q21q26.2) lub t(3;3)(q21;q26.2) – RPN1-EVI1 t(6;9)(p23;q34) – DEK-NUP214 t(v;11)(v;q23) – MLL rearanżacje −5 lub del(5q) −7; abnl(17p) złożony kariotyp |
Rokowanie
Największe szanse mają chorzy z korzystnymi zmianami cytogenetycznymi, bez istotnych chorób współistniejących w wieku <60 lat, u których leczenie indukcyjne szybko doprowadziło do remisji całkowitej.
Całkowitą remisję osiąga 80–90% leczonych w grupie korzystnego ryzyka, 50–80% leczonych w grupie pośredniego I ryzyka, 40–60% leczonych w grupie pośredniego II ryzyka i około 50% leczonych w grupie niekorzystnego ryzyka. Do nawrotu w grupie korzystnego ryzyka u chorych bez przeszczepionego szpiku dochodzi u około 30–40% chorych, a u chorych po przeszczepieniu u około 15–20%. W grupie pośredniego I ryzyka do nawrotu u chorych bez przeszczepienia i po przeszczepieniu dochodzi odpowiednio w 50–60% oraz 20–25% przypadków. W grupie ryzyka pośredniego II wielkości te wynoszą 70–80% bez przeszczepienia i 30–40% po przeszczepieniu. W grupie wysokiego ryzyka nawrót u chorych niepoddanych przeszczepieniu szpiku dotyczy aż 90%, przeszczep redukuje te ryzyko do 40–50%.
Nawroty są najczęstsze w pierwszym roku leczenia, w miarę upływu czasu są coraz mniej prawdopodobne. Zastosowanie samej chemioterapii daje bardzo duże szanse wyleczenia ostrej białaczki promielocytowej oraz AML z t(8;21) i inv(16). W innych sytuacjach chemioterapia bez przeszczepienia szpiku nie daje dużych szans na całkowite wyleczenie. W grupie pośredniego ryzyka chemioterapia i przeszczepienie szpiku pozwala na całkowite wyleczenie aż u 60% chorych. Wyniki w grupie wysokiego ryzyka są złe.
Epidemiologia
Ostra białaczka szpikowa stanowi 80% ostrych białaczek u dorosłych i 25% wszystkich białaczek u dorosłych, ale jest stosunkowo rzadka u dzieci, które częściej chorują na ostrą białaczkę limfoblastyczną. Zapadalność zwiększa się wraz z wiekiem. Ryzyko zachorowania w sześćdziesiątym roku życia wynosi 10/100 000, a w osiemdziesiątym roku życia około 20/100 000.
U dzieci ostra białaczka szpikowa jest rzadsza od ostrej białaczki limfoblastycznej i stanowi 18% wszystkich przypadków białaczki u dzieci. AML u dzieci występuje z częstością 7-8 przypadków na milion dzieci poniżej 15. roku życia. Największa zapadalność u dzieci występuje około 1. roku życia, zapadalność w tej grupie wiekowej wynosi 18,4 przypadków na milion dzieci, następnie spada do 4,3 przypadków na milion w grupie wiekowej dzieci 5-9-letnich, następnie zapadalność znów rośnie do 7,7 przypadków na milion.
Ryzyko zachorowania w Polsce na AML u osób powyżej 18. roku życia wynosi 2,1/100 000.
Mężczyźni chorują znacząco częściej niż kobiety.
Historia

Pierwszy opis choroby jest przypisywany wielu lekarzom. W 1811 roku Peter Cullen opisał przypadek młodej kobiety z gorączką, bólami brzucha i powiększeniem śledziony. Podczas upustów krwi, którym poddawano chorą, zauważył, że jej krew przyjmuje mleczne zabarwienie i konsystencję.
W 1825 roku Alfred Velpeau opisał chorobę, która charakteryzowała się osłabieniem, gorączką, świądem w obrębie skóry brzucha i kamicą moczową. Podczas sekcji zwłok stwierdził znaczne powiększenie wątroby i śledziony oraz zmienioną konsystencję krwi przypominającą kaszę lub ropę. Kolejne opisy podali Jacques Charles Collineau w 1829 roku oraz A. Duplay w 1834 roku. W 1845 roku John Hughes Bennett powiązał stwierdzone zmiany patologiczne z nagromadzeniem leukocytów w organizmie, które określił jako „leukocytemia”. Rudolf Virchow zauważył, że zmieniony wygląd krwi chorych wynika ze zmniejszenia liczby krwinek czerwonych przy jednoczesnym zwiększeniu liczby krwinek białych. W 1847 roku wprowadził on pojęcie białaczki (leukämie), a w 1856 roku wyróżnił białaczkę śledzionową i limfatyczną.
Pierwszym lekarzem, który rozpoznał białaczkę za życia chorego był Henry William Fuller. Wilhelm Ebstein w 1869 roku wyróżnił ostre białaczki, które charakteryzują się szybszym przebiegiem choroby od przewlekłych białaczek. Franz Ernst Christian Neumann w 1869 roku zlokalizował hematopoezę w szpiku kostnym. Wkrótce potem F. Mosler wprowadził biopsję szpiku kostnego do diagnostyki białaczki, która wcześniej była stwierdzana głównie po śmierci. W 1900 roku Otto Naegeli opisał mieloblasty oraz limfoblasty, co dało podstawę do podziału na białaczki szpikowe i limfoblastyczne.
Początkowo ostrą białaczkę szpikową leczono upustami krwi. Następnie do lat 30. XX wieku choroba była leczona poprzez uzupełnianie niedoborów żelaza. W latach 50. próbowano leczyć ostrą białaczkę szpikową za pomocą radioaktywnego fosforu, jednak wyniki leczenia były słabe. Dopiero wprowadzenie pierwszych skutecznych cytostatyków dało nadzieję na skuteczne leczenie choroby. W 1948 roku Sidney Farber wprowadził do leczenia ostrej białaczki limfoblastycznej aminopterynę. Gertrude Elion oraz George Hitchings w latach 1950–1951 opracowali jeden z pierwszych skutecznych leków przeciw ostrej białaczce szpikowej – merkaptopurynę. E. Donnall Thomas w latach 70. wprowadził przeszczepianie szpiku kostnego do leczenia ostrej białaczki szpikowej. W 1976 roku wprowadzono klasyfikację francusko-amerykańsko-brytyjską, a w 2008 klasyfikację opartą na zmianach cytogenetycznych.
Uwagi
Bibliografia
- M.R. O’Donnell, M.S. Tallman, C.N. Abboud, J.K. Altman i inni. Acute myeloid leukemia, version 2.2014. „National Comprehensive Cancer Network”, 2014.
- M.F. Fey, C. Buske. Acute myeloblastic leukaemias in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. „Ann Oncol”. 24 Suppl 6, Oct 2013. DOI: 10.1093/annonc/mdt320. PMID: 23970018.
- Andrzej Szczeklik, Piotr Gajewski: Interna Szczeklika 2014. Kraków: Medycyna Praktyczna, 2014. ISBN 978-83-7430-405-4.
- Vincent T. DeVita, Theodore S. Lawrence, Steven A. Rosenberg: Devita, Hellman & Rosenberg’s Cancer: Principles & Practice of Oncology. Wyd. 8. Lippincott Williams & Wilkins, 2008. ISBN 978-0-7817-7207-5.
- Michael J. Perry: The Chemotherapy source book. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins, 2008.
- Ronald Hoffman, Edward J. Benz Jr., Leslie E. Silberstein, Helen Heslop, Jeffrey Weitz, John Anastasi: Hematology: Diagnosis and Treatment. Elsevier Health Sciences, 2013. ISBN 978-1-4557-7688-7.
- Lalitha Nagarajan: Acute Myelogenous Leukemia: Genetics, Biology and Therapy. Springer Science & Business Media, 2010. ISBN 978-0-387-69259-3.
- Kristian Reckzeh: Deciphering the pathogenesis of acute myeloid leukemia. 2012. ISBN 978-91-86871-99-4.