
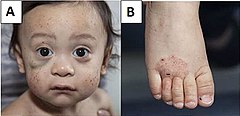
 Zespół Wiskotta-Aldricha: A) Liczne wybroczyny na twarzy i krwiak pod prawym okiem. B) Wyprysk na stopie. | |
| Klasyfikacje | |
| ICD-10 | |
|---|---|
Zespół Wiskotta-Aldricha (ang. Wiskott-Aldrich syndrome, WAS) – rzadki dziedziczny zespół chorobowy, sprzężony z chromosomem X, o typie dziedziczenia recesywnym, na który choruje jedynie płeć męska. Choroba przebiega z zaburzeniami układu odpornościowego. Wystąpienie objawów choroby jest uwarunkowane mutacją genu WAS w locus Xp11.23-p11.22, kodującego białko o tej samej nazwie. Białko WASP jest obecne w komórkach progenitorowych krwi, limfocytach T i B, makrofagach oraz trombocytach, co wskazuje, że jest ono niezbędne do prawidłowego różnicowania się i funkcjonowania tych komórek. Choroba może przebiegać od postaci łagodnych z okresową trombocytopenią, po znaczne zaburzenia odporności. Niezależnie jednak od aktualnego stanu zaawansowania, objawy chorobowe nasilają się wraz z upływem czasu. Występuje z częstością 1:250 000 chłopców. Po raz pierwszy zespół opisany w 1954 przez Aldricha i współpracowników.
Objawy chorobowe
- wyprysk
-
trombocytopenia
- krwawa biegunka
- wybroczyny skórne
- upośledzenie czynności limfocytów B i T powodujące nawracające zakażenia
- zapalenie płuc
- zapalenie oskrzeli
- zapalenie ucha środkowego
- zakażenia skóry
- zakażenia wirusem opryszczki
- hepatosplenomegalia jako wyraz przewlekłego stanu zapalnego.
Osoby chorujący na zespół Wiskotta-Aldricha wykazują zwiększoną skłonność do zachorowań na nowotwory złośliwe, takie jak np. białaczkę i chłoniaki oraz choroby autoimmunologiczne.
Badania dodatkowe
- trombocytopenia poniżej 100 000
- niskie stężenie immunoglobuliny M
- niskie stężenie izohemaglutynin
- prawidłowe stężenia IgG i prawidłowe lub podwyższone IgE i IgA
- biopsja szpiku celem wykluczenia innych schorzeń przebiegających z zaburzeniami odporności (sam zespół Wiskotta-Aldricha nie powoduje charakterystycznych zaburzeń szpiku kostnego)
Stopnie zaawansowania
Jin i współpracownicy w 2004 zaproponowali następującą klasyfikację ciężkości choroby:
- 0,5 trombocytopenia nawracająca
- 1,0 trombocytopenia i małe płytki
- 2,0 trombocytopenia, leczący się wyprysk skóry, sporadyczne infekcje dróg oddechowych
- 3,0 wyprysk skórny i infekcje dróg oddechowych wymagające stosowania antybiotyków
- 4,0 wyprysk skórny wymagający stałego leczenia lub zagrażające życiu infekcje
- 5,0 choroba autoimmunologiczna lub nowotworowa
Leczenie
- przetoczenia koncentratów krwinek płytkowych w przypadku małopłytkowości (trombocytopenii)
- agresywna antybiotykoterapia w przypadku zakażeń
- splenektomia w przypadku nasilonej małopłytkowości
- dożylne podawanie immunoglobulin
- w przypadku powikłań pod postacią chorób autoimmunologicznych zwykle stosuje się glikokortykosterydy ogólnie, winkrystynę i plazmaferezę
- przy zmianach skórnych – sterydy w postaci maści
- przeszczep komórek macierzystych (np. szpiku kostnego)
- terapia genowa (eksperymentalna)
Rokowanie
Przez długi okres zespół był uznawany za chorobę śmiertelną ze średnim okresem przeżycia ok. 4 lat. Obecnie skuteczna terapia zapobiegająca infekcjom i krwawieniu pozwala na relatywnie normalne życie. Terapia przeszczepem komórek macierzystych poprzez przeszczep szpiku kostnego lub krwi pępowinowej, stosowana w najbardziej zaawansowanych przypadkach, umożliwia całkowite wyleczenie i jest skuteczna w 9 na 10 przypadków.
Historia
Zespół został nazwany od Roberta Andersona Aldricha, amerykańskiego pediatry, który opisał chorobę u holendersko-amerykańskiej rodziny w 1954 i Alfreda Wiskotta, niemieckiego pediatry, który jako pierwszy zauważył zespół w 1937. Wiskott opisał podobną chorobę u trzech braci, których siostra nie była chora. W 2006 niemiecka grupa badawcza przeanalizowała te 3 przypadki opisane przez Wiskotta i wysunęli przypuszczenie, że bracia prawdopodobnie posiadali nowo powstałą mutację przesunięcia ramki odczytu w eksonie genu WAS.
Zobacz też
Linki zewnętrzne
- WISKOTT-ALDRICH SYNDROME; WAS w bazie Online Mendelian Inheritance in Man (ang.)